





Abstract
Large outbreaks of tuberculosis (TB) represent a particular threat to disease control because they reflect multiple instances of active transmission. The extent to which long chains of transmission contribute to high TB incidence in London is unknown. We aimed to estimate the contribution of large clusters to the burden of TB in London and identify risk factors.
We identified TB patients resident in London notified between 2010 and 2014, and used 24-locus mycobacterial interspersed repetitive units–variable number tandem repeat strain typing data to classify cases according to molecular cluster size. We used spatial scan statistics to test for spatial clustering and analysed risk factors through multinomial logistic regression.
TB isolates from 7458 patients were included in the analysis. There were 20 large molecular clusters (with n>20 cases), comprising 795 (11%) of all cases; 18 (90%) large clusters exhibited significant spatial clustering. Cases in large clusters were more likely to be UK born (adjusted odds ratio 2.93, 95% CI 2.28–3.77), of black-Caribbean ethnicity (adjusted odds ratio 3.64, 95% CI 2.23–5.94) and have multiple social risk factors (adjusted odds ratio 3.75, 95% CI 1.96–7.16).
Large clusters of cases contribute substantially to the burden of TB in London. Targeting interventions such as screening in deprived areas and social risk groups, including those of black ethnicities and born in the UK, should be a priority for reducing transmission.
Abstract
Large clusters contribute substantially to the burden of tuberculosis in London, indicating ongoing transmission http://ow.ly/3xk23068P6w
Introduction
In countries with low incidence of tuberculosis (TB) such as the UK, highest rates are often found in large cities [1]. The rate of TB in London in 2014, for example, was 30 per 100 000 population compared with 12 per 100 000 in the whole of England [2]. This high incidence has led to the city being described as the “TB capital of Western Europe” [3].
Large outbreaks of TB represent a particular threat to control because they reflect multiple instances of active transmission. Such large outbreaks have occurred previously in London and other large cities [4–9]. However, identification of outbreaks of TB is difficult, as it requires cases resulting from active transmission to be distinguished from those resulting from reactivation of latent disease with absent or limited onward transmission. The extent to which they contribute to the overall disease burden is therefore not known.
Molecular strain typing provides one means of linking cases that may be part of outbreaks. Cases that share a molecular strain type may be linked through transmission and therefore form part of large outbreaks, although they may also reflect common endemic strains. In England, prospective molecular strain typing has been conducted since 2010 using the 24-locus mycobacterial interspersed repetitive units–variable number tandem repeat (MIRU-VNTR) method.
Spatial analyses provide another means of investigating potential links between cases of infectious disease [10]. Tests of spatial clustering, for example, can be used to identify cases that occur closer together in space than would be expected by chance. They can therefore be used in combination with molecular data to assess evidence for recent transmission in investigations of TB clusters.
An analysis of the first 3 years of MIRU-VNTR data in London showed that 46% of cases were part of a molecular cluster and identified risk factors for clustering [11]. It also showed that cluster size ranged from two to 55 cases, and that over half of the clusters had only two cases. However, the study did not determine whether risk factors varied by cluster size or assess spatial clustering.
In this study, we investigated the size and distribution of molecular clusters of TB in London between 2010 and 2014 using routine molecular strain typing and epidemiological data. We aimed to quantify the contribution of large molecular clusters to the burden of TB in the city, describe the characteristics of cases by cluster size and identify risk factors. We also aimed to assess evidence for transmission in large molecular clusters by testing for spatial clustering.
Methods
Study population and data sources
This was a cross-sectional analysis of patients notified with TB between January 1, 2010 and December 31, 2014 resident in London. Data were extracted from the Enhanced Tuberculosis Surveillance (ETS) system, a national online register for real-time case reporting that is run by Public Health England (PHE). This system includes demographic (age, sex, ethnic group, country of birth, time since entry to the UK and occupation) and clinical (site of disease, sputum smear status, history of TB disease and treatment, drug sensitivity, and whether the case spent time as a hospital inpatient) characteristics of patients. It also includes patient residential locations and information on social risk factors for TB (whether the patient has a history of homelessness, or problems with illicit drug or alcohol use). Surveillance data from ETS is routinely matched to the National Tuberculosis Strain Typing Service to provide MIRU-VNTR molecular clustering data.
An estimate of level of social deprivation (the index of multiple deprivation (IMD)) is also included in the ETS system. This is obtained through matching of residential postcodes to Lower Layer Super Output Areas (LSOAs), a geographic hierarchy used in England and Wales each encompassing a mean population of 1500. The IMD is a measure of relative deprivation at the LSOA level in England and is based on seven domains of deprivation: 1) income, 2) employment, 3) crime, 4) living environment, 5) barriers to housing and services, 6) health and disability, and 7) education, skills and training [12]. Low ranks indicate higher levels of deprivation. We converted IMD ranks into London-level deprivation quintiles, with the lowest quintile representing the most deprived areas.
Ethical approval was not required for this study because it was based on PHE routine surveillance data. PHE has Health Research Authority approval to hold and analyse national surveillance data for public health purposes.
Molecular clustering analysis
We categorised cases as unique or part of a molecular cluster and by the number of cases in the molecular cluster. We used the PHE convention for assigning cluster status. Molecular clusters were groups of two or more cases that shared an identical MIRU-VNTR strain type with another case notified in the study region during the study period. Unique cases were individuals whose strain type did not cluster with another case. We excluded cases who did not have an isolate typed by MIRU-VNTR with at least 23 loci and those whose molecular strain type was unique within the study area but shared a molecular strain type with another case in England.
We described the distribution of molecular clusters by size (number of cases in the cluster) and calculated the proportion of cases which were part of clusters. We identified successive cases reported in a molecular cluster using case notification dates and calculated the median and interquartile range (IQR) number of days between successive cases in molecular clusters by cluster size.
In this analysis, we aimed to investigate risk factors for cases belonging to a molecular clusters of different sizes. We therefore categorised cases according to the size of the molecular cluster (not clustered (unique) cases, n=2 cases, n=3–20 cases and n>20 cases). In situations such as this, in which the outcome of interest is a categorical variable, a multinomial logistic regression model can be used [13]. This is an extension of the simple logistic regression model which is used for dichotomous outcomes. Coefficients resulting from the multinomial model are interpreted in a similar way to the odds ratios (ORs) derived from a logistic regression model.
We investigated associations at single-variable analysis and included variables with an association of p<0.2 in the initial multivariable model. A backwards stepwise approach was then used to eliminate variables which did not contribute significantly to produce a final model. LSOA of residence was included as a random effect in models which included the IMD to account for the hierarchical level at which this variable was measured. Social risk factors were considered separately, and as a cumulative count of these risk factors at single-variable analysis and as a count at multivariable analysis.
Spatial clustering analysis
We used spatial scan statistics to assess spatial clustering within molecular clusters, implemented using SaTScan software (www.satscan.org). We tested the hypothesis that cases in large molecular clusters (n>20 cases) were closer together in space than the underlying spatial distribution of TB cases. For each large molecular cluster, we therefore performed a spatial scan under the Bernoulli (case–control) model, using the locations of all other TB cases as controls.
We aimed to identify areas with evidence of local transmission and therefore set the maximum radius of the spatial window at 5 km and identified clusters with a p-value of <0.05 which encompassed at least 10 cases. We plotted the locations of significant spatial clusters for each molecular cluster overlaid on a smoothed incidence map of the relative distribution of the given molecular cluster compared with all other TB cases. These maps were generated through kernel density estimation using a Gaussian kernel of bandwidth 5 km.
Statistical analyses were performed using R version 3.2.3 (www.r-project.org).
Results
Between 2010 and 2014, a total of 15 670 cases of TB were notified in London. Of these, 8148 (52%) cases were successfully typed by MIRU-VNTR with at least 23 loci defined, whilst 6241 (40%) were not culture confirmed and 1281 (8%) were not typed, and therefore excluded from this analysis (figure 1). A further 690 cases were also excluded because they clustered only with cases that were not resident within the study area. This study therefore included 7458 TB cases with a molecular strain type, of which 4129 (55%) were part of 996 molecular clusters and 3329 (45%) had a unique strain.
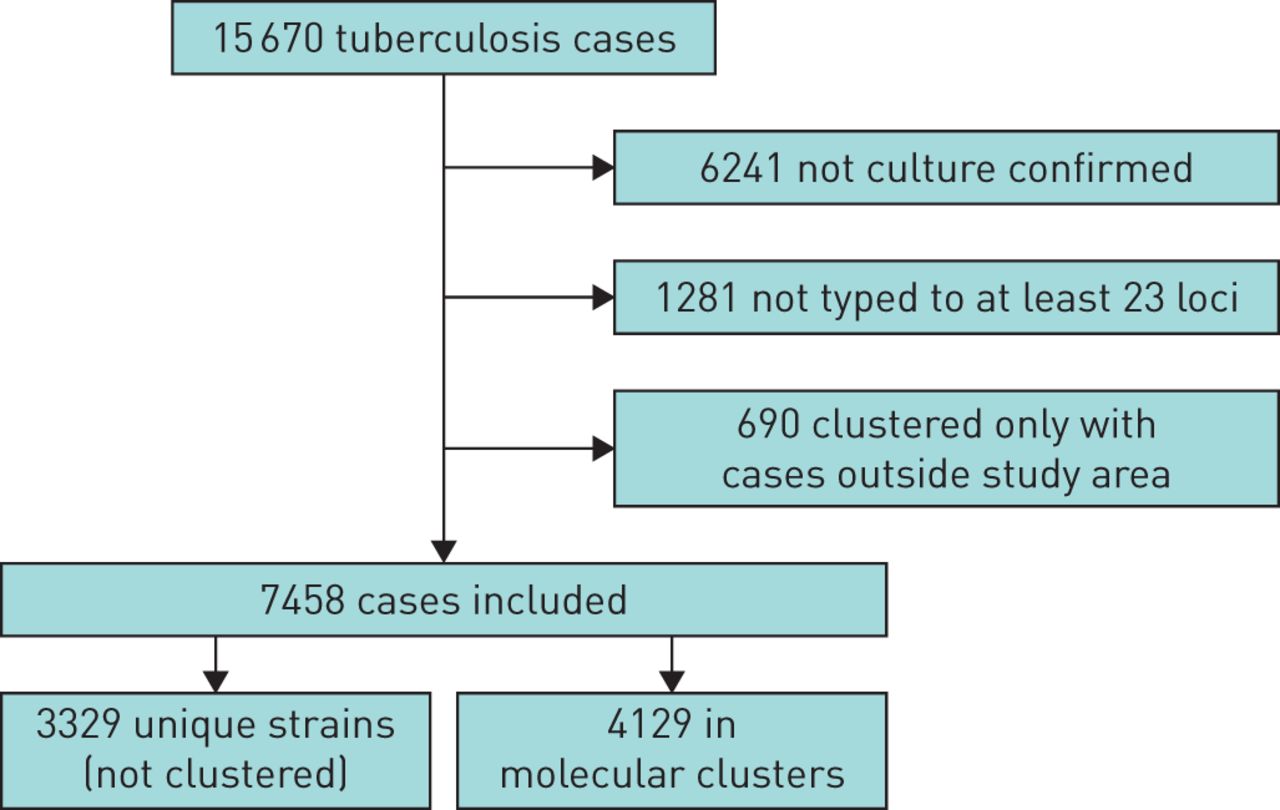
Cases included in analysis of molecular clusters of tuberculosis in London (2010–2014).
Cluster size and time between cases
Cluster size ranged from two to 102 cases, with a median of n=2 cases. There were 20 clusters with n>20 cases, including 795 (11%) of all cases (table 1). Over half of the clusters (522 (53%)) comprised pairs of cases, but a larger proportion of cases were in the 454 clusters of n=3–20 cases (2290 (31% of cases)).
Distribution of tuberculosis cases and molecular clusters in London (2010–2014), by cluster size
Successive cases in clusters were defined using notification dates. Figure 2 displays the distribution of median intervals between successive cases in each cluster by cluster size. Overall, the median (IQR) time between successive cases in a cluster was 114 (32–323) days. For cases in clusters of n>20 cases the median (IQR) time was 23 (8–54) days, for cases in clusters of n=3–20 cases it was 149 (54–335) days) and for clusters of n=2 cases it was 406 (162–752) days.
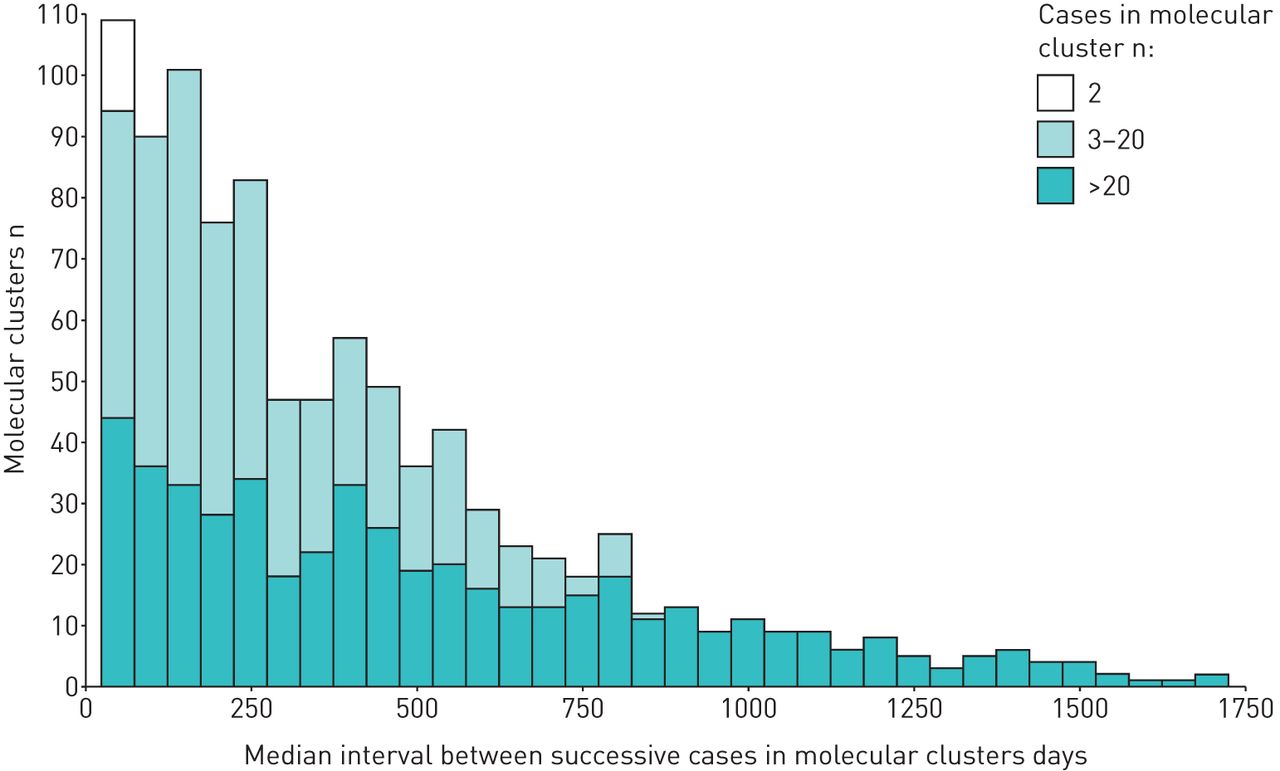
Median interval between successive tuberculosis cases in molecular clusters in London (2010–2014), by cluster size.
Factors associated with large clusters
Baseline characteristics of TB cases according to the number of cases in the cluster are shown in table 2 and results of the single-variable multinomial logistic regression analysis are presented in table 3.
Baseline characteristics of tuberculosis cases in molecular clusters of different sizes in London (2010–2014)
Single-variable multinomial logistic regression analysis for risk factors associated with tuberculosis cases in molecular clusters of different sizes in London (2010–2014)
For each exposure, an OR was calculated for each of the three cluster size outcomes (n=2, 3–20 and >20 cases), with cases not in a cluster representing the comparison group. For example, the unadjusted ORs for being born in the UK were 4.11 (for cases in clusters of n>20 cases), 2.55 (for cases in clusters of n=3–20 cases) and 1.77 (for cases in clusters of n=2 cases). This means that the odds of cases being in the largest clusters versus not being in a cluster for those born in the UK were 4.11 times that of those not born in the UK. Similarly, the odds of cases being in a cluster of n=2–20 cases compared with not being in a cluster for those born in the UK were 2.55 times that of those not born in the UK; and the odds of cases being in a cluster of n=2 cases compared with not being in a cluster for those born in the UK were 1.77 times those not born in the UK.
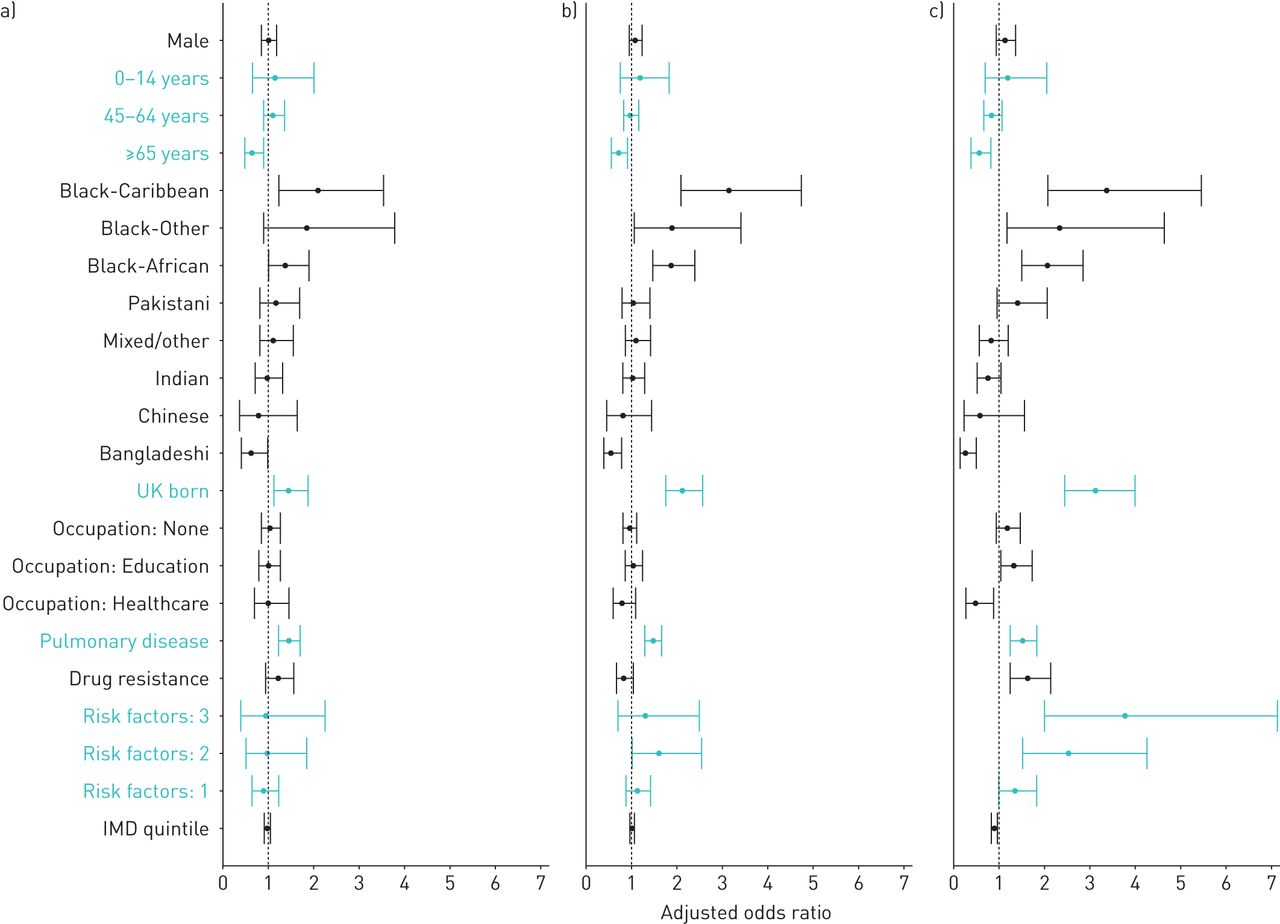
Factors included in the final multivariable model were sex, age, ethnicity, place of birth, occupation, site of disease, drug resistance, number of social risk factors and IMD (table 4 and figure 3). Cases in the oldest age group (≥65 years) had an adjusted OR (aOR) of 0.52 (95% CI 0.35–0.78); aOR for being born in the UK was 2.93 (95% CI 2.28–3.77). The association between black ethnic groups and larger cluster size was maintained (aOR black-Caribbean ethnicity 3.64, 95% CI 2.23–5.94), whilst the only ethnic group with significantly lower risk than the white population was Bangladeshi (aOR 0.26, 95% CI 0.13–0.50). Students and those working in education had an increased adjusted odds of being in large clusters (aOR 1.31, 95% CI 1.01–1.70), and those working in healthcare had a decreased adjusted odds (aOR 0.47, 95% CI 0.25–0.87).
Multivariable multinomial logistic regression analysis for risk factors associated with tuberculosis cases in molecular clusters of different sizes in London (2010–2014), adjusted for random effects of Lower Layer Super Output Area (LSOA)

Forest plot of adjusted odds ratios (with 95% confidence intervals) from multivariable multinomial logistic regression analysis (table 4), by number of cases in molecular cluster: a) n=2, b) n=3–20 and c) n>20 cases.
Social risk factors were included in the final model as a count and there was a trend of increased odds with increased number of risk factors, although confidence intervals overlapped (aOR three risk factors 3.75, 95% CI 1.96–7.16; aOR four risk factors 16.64, 95% CI 1.98–139.88). Deprivation was also independently associated with being in a large cluster; the aOR was 0.90 (95% CI 0.83–0.97) for increased IMD quintile and therefore decreased deprivation level.
Black-Caribbean ethnicity, being born in the UK and pulmonary disease were the only factors that also had significantly elevated odds for clusters of n=2 or n=3–20 cases.
Spatial clusters of cases in large molecular clusters
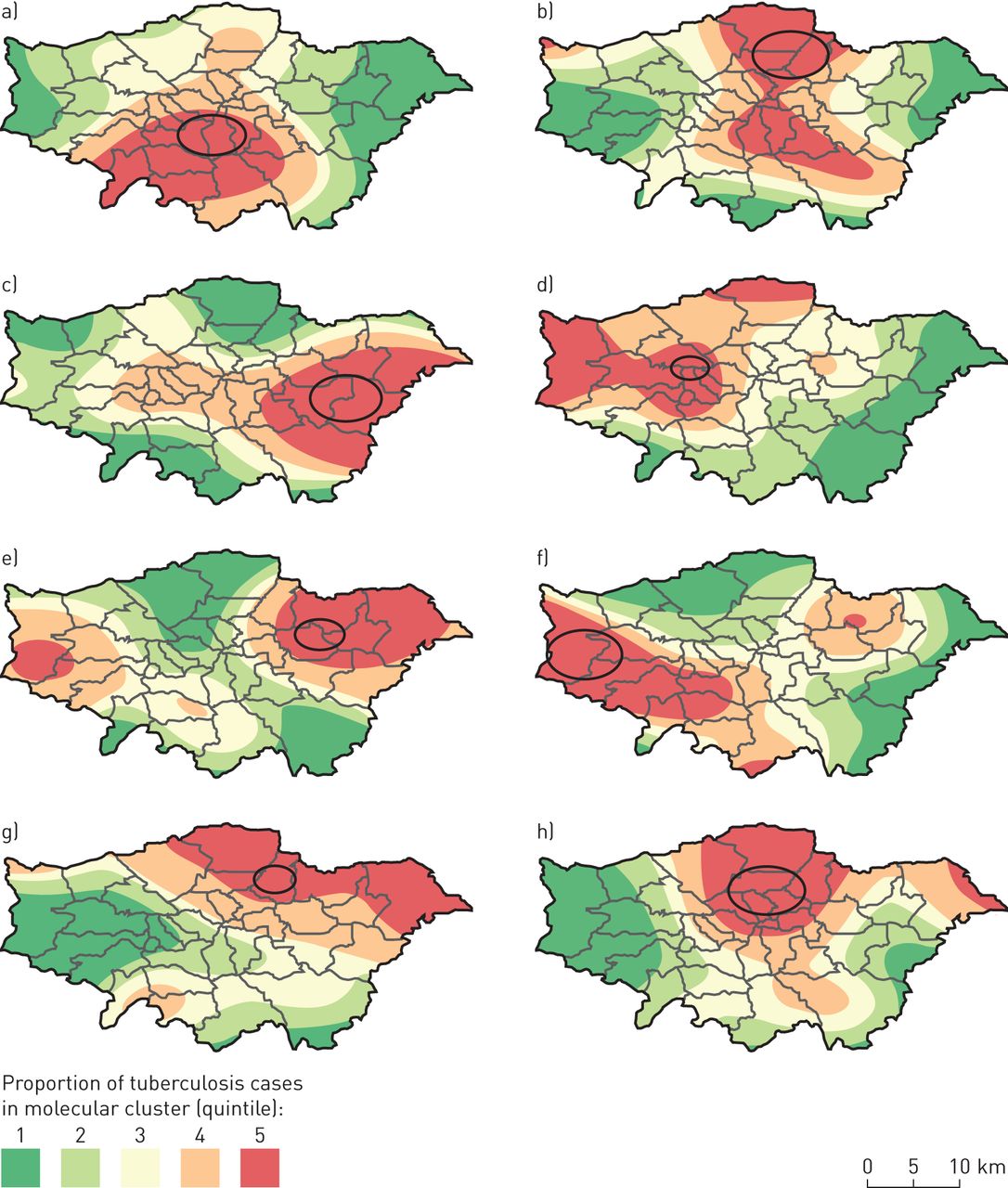
We used SaTScan to test for spatial clustering in the 20 molecular clusters that had n>20 cases. A total of 25 significant spatial clusters (p<0.05) were identified, with at least one significant spatial cluster in 18 (90%) of the molecular clusters, and eight of the spatial clusters included more than 10 cases. These clusters tended to be located in more deprived areas; the median IMD rank of the 4970 LSOAs within London for areas within the clusters was 1110 compared with 2538.5 for areas not in clusters.
The locations of the eight spatial clusters, overlaid on smoothed incidence maps, are shown in figure 4.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a–h) Locations of significant spatial clusters of cases within eight molecular clusters of tuberculosis (TB) in London (2010–2014), overlaid on smoothed incidence maps. Ovals represent areas of significant spatial clustering (p<0.05) with more than 10 cases of the given molecular cluster compared with the general distribution of TB cases. The proportions of cases in molecular clusters compared with all other TB cases are represented through kernel density estimation (bandwidth 5 km).
Discussion
In this study, we present results from the first 5 years of routine molecular strain typing of TB by MIRU-VNTR in London. There were 20 molecular clusters that had n>20 cases of TB notified between 2010 and 2014. These clusters accounted for 795 (11%) of all typed cases notified during this period, and cases in large clusters also tended to occur closer together in space and time. One of the molecular clusters described in this study is part of a known outbreak of isoniazid-resistant disease that was first identified in 1999 [5], but this is the first analysis to suggest that multiple similar outbreaks may be ongoing.
Cases in large molecular clusters were more likely to have multiple social risk factors, be of black ethnicities, born in the UK, have pulmonary and drug-resistant disease, and live in more deprived areas of London. Small clusters (pairs of cases) and those of intermediate size (n=3–20 cases) were associated with black-Caribbean ethnicity, being born in the UK and pulmonary disease. There was also some association between large clusters and occupation. Large clusters were more likely to include students and those involved in education, which may suggest that outbreaks in schools and universities can spread widely in these settings or through extensive social networks involving students. However, they were less likely to involve healthcare workers. This indicates that there was limited nosocomial transmission and that when transmission involving a healthcare worker did occur it was usually an isolated incident rather than part of a large outbreak.
The majority of large molecular clusters exhibited significant spatial clustering, indicating likely transmission within London. Spatial clusters tended to be in more deprived areas and the IMD was independently associated with being in a large molecular cluster, after accounting for individual risk factors. Studies in other settings, including Lima, Peru [14], northern England [15], Tokyo, Japan [16] and the USA [17], have also investigated TB clustering using molecular and spatial data. Various methods have been used to assess spatial clustering, but all have also identified areas of likely localised TB transmission or “hotspots”.
Previous analyses of MIRU-VNTR strain typing and surveillance data in various settings have sought to determine the proportion of cases that were part of a molecular cluster or to identify risk factors for clustering [11, 18–25]. A systematic review of 27 articles found that clustering estimates ranged from 0% to 63% [26], whilst the clustering proportion in the first 3 years of routine molecular strain typing in London was 46% [11]. This indicates that the rate of molecular clustering identified in our analysis (56%) was relatively high and that it has risen with inclusion of more years of data.
A strength of this study was that it was based on routine surveillance data and therefore included all cases of TB in London that were successfully typed by MIRU-VNTR to at least 23 loci over a 5-year period. There was a low level of missing information in the variables used in the risk factor analysis (table 2). As a result it provides a good representation of the population of TB cases in the city. The study adhered to the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines for reporting of cross-sectional studies [27].
Another strength was the use of multinomial logistic regression to identify risk factors. The advantage of this method was that it allowed associations to be assessed for different sizes of molecular cluster. This is important because larger clusters have more implications for TB control. It therefore extended the previous analysis of risk factors for clustering, which used a binary outcome that was not stratified by cluster size. The combination of molecular with spatial clustering analyses was a further advantage of this study, as it provided further evidence for transmission in some large molecular clusters. It also has practical application, as it could be used to prioritise clusters for further investigation.
A limitation of this study is that we had to restrict our analysis to cases of TB which had been typed by MIRU-VNTR for at least 23 loci. We therefore excluded 7522 cases that were not culture confirmed or typed. This will have resulted in misclassification of some cases as unclustered which did not have a unique strain and therefore underestimated the number of cases in some clusters. A second limitation was that we considered the temporal distribution of cases in molecular clusters by examining the median interval between case notification dates, which are a proxy for dates of onset. This provided some evidence that cases in larger molecular clusters occurred closer together in time than those in smaller clusters. However, a true estimate of serial intervals to assess this robustly would require ascertainment of epidemiological links between cases to establish chains of transmission. Our analysis was also limited because we were unable to assess the importance of other potential factors affecting TB transmission which are not currently collected in surveillance data. HIV status, for example, is not routinely collected, although all TB patients are offered HIV testing and it was taken up by 98% patients in London in 2014 [28]. Modelling estimates show that HIV co-infection in TB patients in England is relatively low, at just 3.4% in 2014 (personal communication, PHE National Infection Service).
The results of this study have implications for the control of TB in London and other high-incidence cities. Targeting interventions to deprived areas should be a priority for reducing transmission, whilst efforts should also be made to raise awareness of the disease amongst at-risk groups, such as those of black ethnicities born in the UK. An example of such an intervention is the “Find and Treat” mobile radiography unit which actively screens for cases in vulnerable populations in London and provides support to help patients complete treatment [29]. Continued support for this service is therefore a key component of TB control in London. Our results also imply that detailed investigations of molecular clusters could be beneficial in preventing large chains of transmission through interventions such as contact tracing and screening. We recommend incorporating routine spatial clustering analysis to assist with prioritising clusters for further investigation, as use of simple thresholds has previously been ineffective in making these decisions [30, 31].
Future work arising from this study could aim to identify which of the components of the IMD may be contributing to TB transmission. This would be useful to inform environmental and housing interventions, such as improving ventilation and reducing overcrowding. More work is also required to determine if the associations observed in clusters of different sizes could be used to predict whether cases in small clusters are likely to form larger clusters. Finally, results from whole genome sequencing of TB isolates when routinely available should add further resolution to networks suggested by molecular clusters. This could provide evidence to support or refute transmission in some instances and direct the focus of intensive investigations [32].
In conclusion, this study shows that large molecular clusters contribute substantially to the burden of TB in London. The results highlight the continued importance of preventing long chains of transmission in order to eliminate TB as a public health problem in large cities.
Acknowledgements
We would like to thank the TB specialist nurses and National Tuberculosis Strain Typing Service for collecting surveillance data and undertaking molecular typing, which enabled us to carry out this study.
- Received September 12, 2016.
- Accepted November 6, 2016.
- Copyright ©ERS 2017.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










