





Abstract
This multicentre, randomised, double-blind, placebo-controlled, two-period crossover study assessed the effect of umeclidinium/vilanterol (UMEC/VI) on exercise capacity in patients with chronic obstructive pulmonary disease (COPD) using the endurance shuttle walk test (ESWT).
Patients were randomised 1:1 to one of two treatment sequences: 1) UMEC/VI 62.5/25 µg followed by placebo or 2) placebo followed by UMEC/VI 62.5/25 µg. Each treatment was taken once daily for 12 weeks. The primary end-point was 3-h post-dose exercise endurance time (EET) at week 12. Secondary end-points included trough forced expiratory volume in 1 s (FEV1) and 3-h post-dose functional residual capacity (FRC), both at week 12. COPD Assessment Test (CAT) score at week 12 was also assessed.
UMEC/VI treatment did not result in a statistically significant improvement in EET change from baseline at week 12 versus placebo (p=0.790). However, improvements were observed in trough FEV1 (206 mL, 95% CI 167–246), 3-h post-dose FRC (−346 mL, 95% CI −487 to −204) and CAT score (−1.07 units, 95% CI −2.09 to −0.05) versus placebo at week 12.
UMEC/VI did not result in improvements in EET at week 12 versus placebo, despite improvements in measures of lung function, hyperinflation and health status.
Abstract
Umeclidinium/vilanterol: no significant improvements in exercise endurance in COPD versus placebo after 12 weeks http://ow.ly/tc9i30gudVc
Introduction
Chronic obstructive pulmonary disease (COPD) is a common disease characterised by persistent respiratory symptoms and airflow obstruction [1]; reduced exercise capacity and/or physical activity often manifest as a result of this obstruction [2–5]. Reduced exercise capacity can have a major impact on patients' health-related quality of life and its improvement is a key goal of COPD disease management [1]. As such, various exercise testing protocols have been developed to evaluate exercise capacity in COPD, including progressive incremental exercise tests and constant work rate exercise (CWRE) tests [6, 7].
The endurance shuttle walk test (ESWT) is a CWRE test conducted on the same flat 10-m course as the incremental shuttle walk test at a speed dictated by an auditory signal [8, 9]. One notable advantage of the ESWT is that it requires less equipment and technical expertise compared with laboratory-based tests [8]. Additionally, the ESWT has been shown to detect functional improvements in response to pulmonary rehabilitation and bronchodilator pharmacotherapy in patients with COPD in a single-centre setting [10–12]. A minimum clinically important difference (MCID) in exercise capacity as measured by the ESWT of 45–85 s (13–15% of the baseline value) in response to a bronchodilator has been validated in previous studies [13–15].
In two previous multicentre studies, the ESWT was used to evaluate the effect of the combination of the long-acting muscarinic antagonist, umeclidinium (UMEC), and the long-acting β2-agonist, vilanterol (VI), on exercise capacity in patients with COPD [16]. Both studies demonstrated that treatment with UMEC/VI (once-daily 62.5/25 or 125/25 µg) resulted in similar improvements from baseline in 3-h post-dose exercise endurance time (EET) over 12 weeks. Consistent improvements in lung function and hyperinflation were observed versus placebo; however, a pronounced placebo effect for EET was observed in one of the studies, possibly confounding the overall interpretation of the results [16].
The aim of this study was to further assess the effect of once-daily UMEC/VI 62.5/25 µg on exercise capacity (as measured by the ESWT) versus placebo in patients with COPD and to evaluate any placebo effect. In addition, the effect of UMEC/VI on measures of lung function and hyperinflation was also assessed.
Material and methods
Study design
This was a single-country (USA), multicentre, randomised, double-blind, placebo-controlled, two-period, complete block design crossover study (ClinicalTrials.gov identifier NCT02275052; GSK identifier 201317), performed between January 2015 and June 2016.
Written informed consent was obtained from all study participants. The study protocol was approved by a national, regional or investigational centre ethics committee or institutional review board, in accordance with Good Clinical Practice. This study was conducted in accordance with Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki (2008).
Patients
Patients eligible for inclusion at screening were ≥40 years of age (male or female) with a diagnosis of COPD [17]; were current or ex-smokers with a smoking history of ≥10 pack-years; had a forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) ratio of <0.70 and a post-bronchodilator FEV1 30–70% of predicted normal; had a resting functional residual capacity (FRC) ≥120% of predicted normal; and had a score ≥2 on the modified Medical Research Council Dyspnoea scale.
Key exclusion criteria at screening were current diagnosis of asthma or other clinically significant respiratory disorder; any other condition or disability that, in the investigator's opinion, would affect the ability to perform the exercise test; unstable or life-threatening cardiac disease or clinically significant ECG readings; COPD exacerbation requiring hospitalisation during the 12 weeks prior to screening; lung volume reduction surgery; oxygen therapy use >12 h·day−1; or participation in the two previous studies conducted to evaluate the effect of UMEC/VI on EET using the ESWT [16]. Patients who met the eligibility criteria at screening completed a run-in period of 11–25 days.
Patients were eligible for randomisation if they had no moderate or severe exacerbations (defined as those requiring antibiotics, systemic corticosteroids or hospitalisation); had no lower respiratory tract infections; and, if using inhaled corticosteroid, had maintained a stable dose equivalent to fluticasone propionate ≤1000 μg·day−1 during the run-in period. Additionally, during the run-in period and at randomisation, patients were required to: demonstrate the ability to properly perform the ESWT without the need for supplemental oxygen; have EETs for the ESWT of ≤15 min that did not vary by >2 min; and have arterial oxygen saturation of ≥85% during the ESWT.
Patient compliance with the study treatment was assessed by reviewing the inhaler's dose counter. Patients who were not compliant with study drug regimen were not excluded from the study, but were re-educated on proper dosing as per the protocol.
Treatments and randomisation
Patients were randomised 1:1 to receive, once daily via the ELLIPTA dry powder inhaler (ELLIPTA is owned by or licensed to the GSK group of companies), either UMEC/VI 62.5/25 µg for 12 weeks followed by placebo for 12 weeks or placebo for 12 weeks followed by UMEC/VI 62.5/25 µg for 12 weeks. The patients received the treatments in two sequences, which were separated by a washout period of 12–17 days to enable washout of the effects of UMEC/VI on the EET and lung function measures. During the washout period, short-acting anticholinergic medications and salbutamol were permitted as required.
Patients were randomised using interactive voice technology and a randomisation schedule generated by Pharmaceutical Product Development (Wilmington, NC, USA).
There were no protocol-defined withdrawal criteria. However, patients were withdrawn from the study if they had a positive pregnancy test or more than one moderate/severe exacerbation.
Outcomes and assessments
The primary end-point was 3-h post-dose EET (as measured using the ESWT) at week 12. Secondary end-points were trough FEV1 at week 12 (defined as the mean of FEV1 values obtained 23 and 24 h post-dose on the previous day), 3-h post-dose FRC at week 12 and 3-h post-dose inspiratory capacity at week 12. Other efficacy end-points included post-dose EET and trough FEV1 at day 2 and week 6, percentage change from baseline in 3-h post-dose EET, trough FVC at week 12, trough FRC at week 12, 3-h post-dose FRC at day 2, trough and 3-h post-dose residual volume at week 12, trough inspiratory capacity at week 12, 3-h post-dose inspiratory capacity at day 2, and COPD Assessment Test (CAT) score [18] at week 12, for evaluation of health status. The 3-h post-dose lung volume measurements were performed directly before the ESWT.
Safety assessments included monitoring of adverse events and COPD exacerbations. A moderate exacerbation was defined as a worsening of COPD symptoms requiring the use of antibiotics and/or systemic corticosteroids; a severe exacerbation was defined as a worsening of COPD symptoms requiring hospitalisation.
Statistical analysis
The study was powered assuming an EET residual within-patient standard deviation of 169 s. Using this assumption, it was calculated that a study with 126 evaluable patients would have 90% power to detect a 70 s difference in EET between UMEC/VI and placebo at the two-sided 5% significance level [15].
To account for multiplicity, a step-down closed testing procedure was applied whereby inference for UMEC/VI versus placebo was dependent upon statistical significance having first been achieved for the previous end-point. The hierarchy consisted of the comparison of UMEC/VI versus placebo performed in the following order: 3-h post-dose EET at week 12, trough FEV1 at week 12, 3-h post-dose FRC at week 12 and 3-h post-dose inspiratory capacity at week 12.
The intent-to-treat (ITT) population comprised all patients randomised to treatment and included those who discontinued treatment.
The primary mixed model repeated measures analysis was conducted on the ITT population including data collected after treatment discontinuation. A supportive analysis was conducted using the ITT population including on-treatment data only (data collected after treatment discontinuation were not included).
A post hoc exploratory analysis was carried out to assess the variability of a subject's EET measurements. The variability of the EET data between the last visit in the run-in period and the visit at randomisation was determined by calculating the interclass coefficient (ICC) for each study centre. ICC was calculated using the between- and within-patient variances from a mixed model with fixed effects of visit, smoking status and patient fitted as random effects.
Correlation between the improvements in EET with UMEC/VI compared with placebo and baseline FEV1, inspiratory capacity and FRC was assessed graphically and using Spearman's rank correlation coefficient.
Results
Demographics
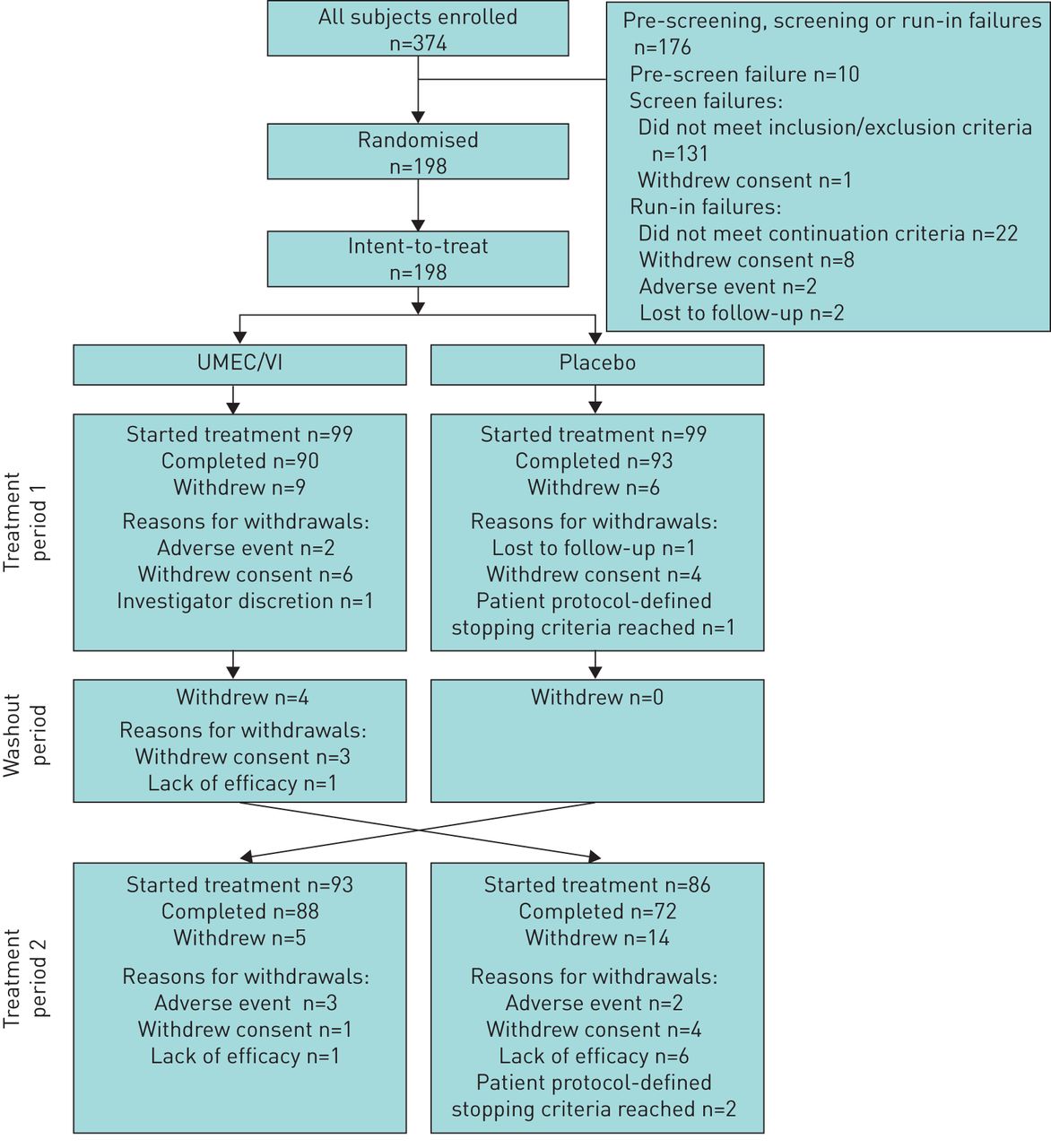
Of the 374 patients enrolled, 198 were randomised to treatment and included in the ITT population (figure 1). A total of 183 (92%) and 160 (81%) patients completed the first and second treatment periods, respectively (figure 1). In total, 15 (8%) patients withdrew during the first treatment period, four (2%) patients withdrew during the washout period and 19 (10%) patients withdrew during the second treatment period. The reasons for withdrawals are shown in figure 1. Baseline demographics and characteristics are presented in table 1. Overall, the majority of patients (64%) were current smokers with a diagnosis of symptomatic moderate-to-severe COPD (Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 2–3 and GOLD categories B and D) (table 1).

Summary of patient disposition. UMEC/VI: umeclidinium/vilanterol.
Demographics and baseline characteristics (intent-to-treat population)
Efficacy
Exercise endurance
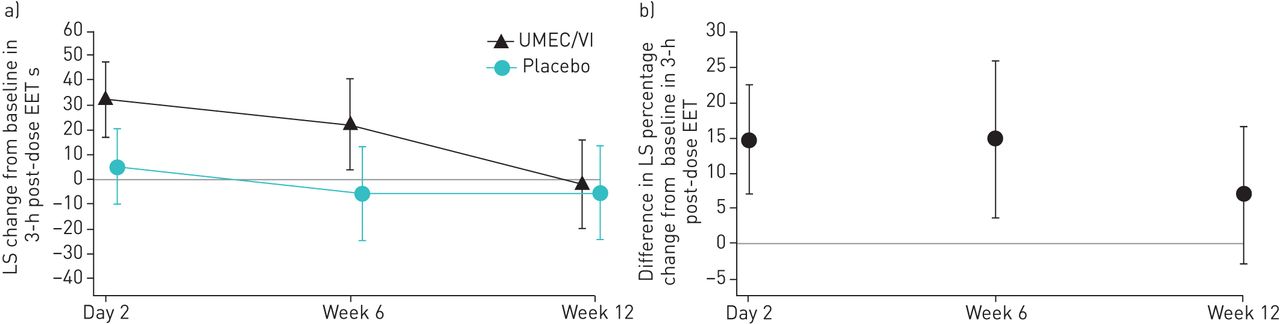
For the primary end-point, UMEC/VI did not result in a statistically significant improvement in EET change from baseline at week 12 compared with placebo (3.3 s, 95% CI −21.1–27.7; p=0.790) (table 2 and figure 2a). Improvements in EET with UMEC/VI compared with placebo were observed at earlier evaluations obtained at day 2 and week 6 (table 2 and figure 2a). No correlation existed between the improvements in EET with UMEC/VI compared with placebo and baseline FEV1, inspiratory capacity or FRC (supplementary figure S2).
Exercise capacity as measured by the exercise endurance test at day 2, week 6 and week 12 (intent-to-treat population including data collected following discontinuation from study treatment)

a) Least squares (LS) mean change (95% CI) from baseline in 3-h post-dose exercise endurance time (EET) (intent-to-treat (ITT) population including data collected following discontinuation from study treatment). b) Percentage change from baseline in 3-h post-dose EET (ITT population including on-treatment data only). UMEC/VI: umeclidinium/vilanterol.
The difference in least squares mean percentage change from baseline in 3-h post-dose EET in the UMEC/VI group compared with the placebo group was 6.87% (95% CI −2.86–16.61) at week 12 (figure 2b). At day 2 and week 6, the corresponding differences in least squares mean percentage change from baseline were 14.67% (95% CI 6.97–22.36) and 14.72% (95% CI 3.59–25.84), respectively (figure 2b).
The ICC values for the variability in EET measurements indicated a high similarity of results between the run-in period and randomisation visits for an individual patient (0.84–0.98) (supplementary table S1).
Trough FEV1
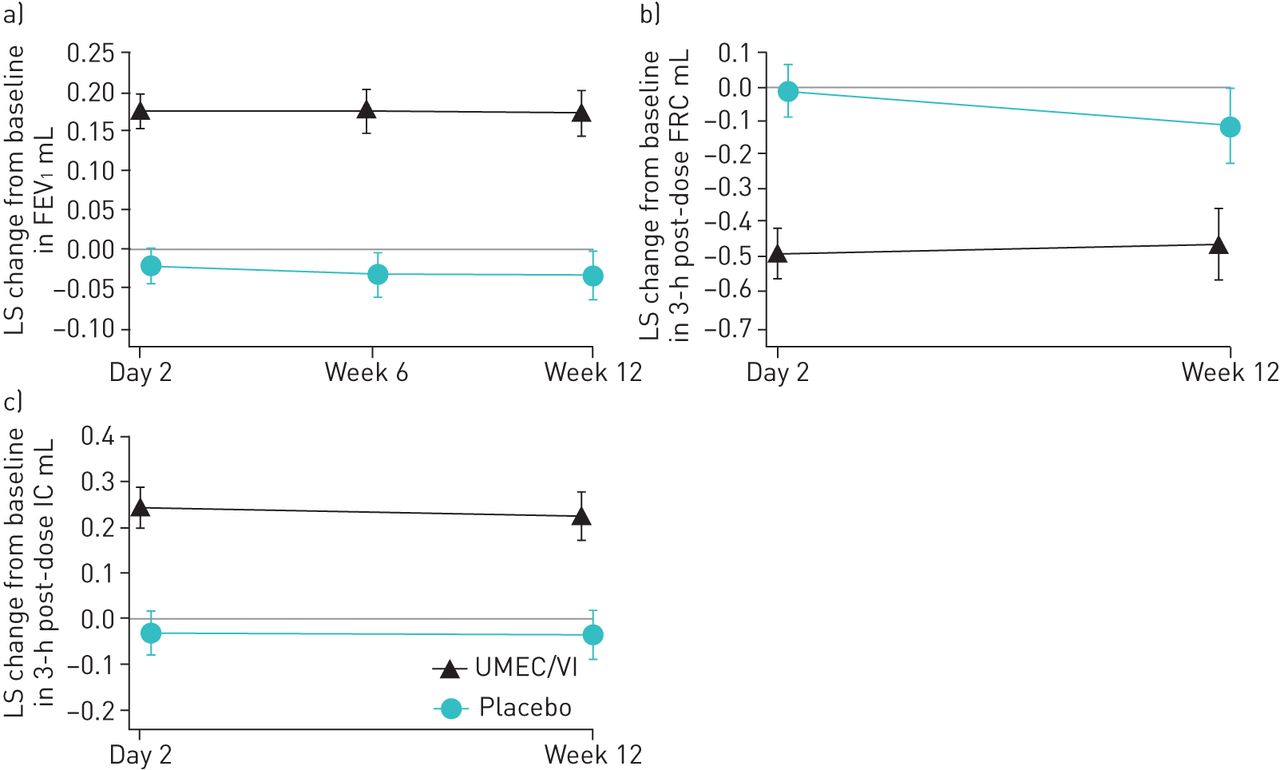
As there was no statistically significant difference on the primary end-point, no inferences to the significance of subsequent outcomes in the testing hierarchy could be made and these data will be presented directionally. For trough FEV1, treatment with UMEC/VI resulted in improvements compared with placebo at week 12 (206 mL, 95% CI 167–246) and at earlier evaluations (figure 3a).

{kind=link}
{kind=link}
{kind=link}
Least squares (LS) mean change (95% CI) from baseline in a) trough forced expiratory volume in 1 s (FEV1), b) 3-h post-dose functional residual capacity (FRC) (intent-to-treat population including data collected following discontinuation from study treatment) and c) 3-h post-dose inspiratory capacity (IC). UMEC/VI: umeclidinium/vilanterol.
Lung volume
Treatment with UMEC/VI resulted in improvements in 3-h post-dose FRC (−346 mL, 95% CI −487 to −204) (figure 3b) and 3-h post-dose inspiratory capacity (259 mL, 95% CI 194–324) (figure 3c) compared with placebo at week 12. Similar results were observed at day 2 (figure 3b and c).
Other end-points
Treatment with UMEC/VI also resulted in improvements in the CAT score compared with placebo at week 12 (−1.07 units, 95% CI −2.09 to −0.05).
Supporting analyses
The results for EET, trough FEV1, FVC, FRC, residual volume and inspiratory capacity from the ITT population that included on-treatment data only were similar to the results obtained using the ITT population that included off-treatment data (supplementary tables S2 and S3, and supplementary figures S3 and S4).
Safety
The incidence of adverse events was similar between the UMEC/VI group (21%) and the placebo group (23%). The most frequently reported adverse events were nausea, headache and sinusitis (1% in the UMEC/VI group and 2% in the placebo group for each), and upper respiratory tract infection (2% in the UMEC/VI group and 1% in the placebo group).
On-treatment serious adverse events were reported by six (3%) patients in the UMEC/VI group and four (2%) patients in the placebo group. In the UMEC/VI group, four patients each experienced one serious event: congestive heart failure, gastroenteritis, cholecystitis and anaphylactic shock. Two patients experienced multiple serious events: gastrointestinal haemorrhage, pulmonary embolism and atrial thrombosis were reported for one patient, and COPD exacerbation and pneumonia were reported for another patient. In the placebo group, two patients each experienced one serious event: COPD exacerbation and Leriche's syndrome. Two patients experienced multiple serious events: COPD exacerbation and pneumonia were reported for one patient, and COPD exacerbation and cellulitis were reported for another patient.
No fatal serious adverse events occurred during the study. However, three post-treatment deaths were reported. In patients who had received UMEC/VI as their previous treatment, one death was attributed to a stage IV lung adenocarcinoma with metastases to the bone and the cause of the other death was unknown. In the placebo group, one death was attributed to a sarcoma and was not considered by the investigator to be related to the study medication.
Discussion
In two previously reported studies using the ESWT to evaluate the impact of UMEC/VI on EET, UMEC/VI resulted in similar, clinically important [15] improvements in EET from baseline over 12 weeks [16]. However, the improvements in EET compared with placebo were not consistent between the studies. In one study, although initial improvements in EET in the UMEC/VI 62.5/25 µg group compared with placebo were observed at day 2, these were not maintained at week 6 or week 12 [16]. This was attributed to an increase in EET in the placebo group over the 12-week treatment period [16]. The increase was noted early in the study and investigated to determine if it was due to various factors, including a learning effect, or differences in study populations, study methodologies or the execution of tests between patient centres. While the study population was less reversible to both salbutamol and ipratropium plus salbutamol than the study in which a placebo effect was not observed, and included fewer patients who were able to walk at the top level of the endurance test employed, there was no definitive explanation.
In this randomised, double-blind, placebo-controlled, two-period crossover study, the change from baseline in EET in the placebo group was consistent over the study period. The lack of a statistically significant difference between UMEC/VI and placebo was due to a reduced EET response throughout the study, but particularly at week 12 compared with day 2 and week 6. This contrasts with previous studies, in which a greater and consistent improvement from baseline in EET was observed with UMEC/VI over time (58.6–69.1 s at week 12) [16]. The differences in least squares mean percentage change from baseline with UMEC/VI versus placebo in the current study were 15%, 15% and 7% at day 2, week 6 and week 12, respectively. These differences were greater than the reported MCID in EET (13–15% change from baseline [15]) at day 2 and week 6, but not at week 12. It is unclear why the change from baseline in EET with UMEC/VI decreased at week 12 compared with the earlier time-points. It is likely not related to the bronchodilator activity of UMEC/VI, as improvements in lung function and lung hyperinflation measures with UMEC/VI over placebo were maintained over 12 weeks and were consistent with the findings from the previous two studies [16]. During exercise, ventilation increases and expiratory time diminishes, causing dynamic hyperinflation, which results in the exertional dyspnoea and reduced exercise capacity associated with COPD [19–21]. Why the marked and sustained improvements in lung hyperinflation did not translate into improvements in EET is unclear, but may be due to the complex and multifactorial processes involved in walking. It has previously been reported that static hyperinflation is less useful than dynamic hyperinflation in predicting improvements in exercise tolerance [20, 22, 23]. In this study, the large ICC at each study site indicated a high degree of similarity in EET measurements between the run-in period and randomisation visits. A way of reducing between-subject variability should be identified for future exercise studies.
In the BRIGHT study, significant improvements in EET were observed with indacaterol/glycopyrronium 110/50 µg versus placebo after 21 days [24]. Additionally, similar improvements in lung function were observed at day 21 compared with those observed throughout the treatment period in the current study. However, it should be noted that there were methodological differences between the BRIGHT study and the current study, e.g. in the BRIGHT study, cycle ergometry rather than ESWT was used to assess EET, which may have affected the results. Additionally, treatment effects were observed over 21 days in the BRIGHT study compared with 12 weeks in the current study. As the BRIGHT study assessed short-term improvement in EET and the current study did not detect improvements after 6 weeks, it is still uncertain which exercise testing methodology is most appropriate for the long-term assessment of EET. Based on the previous two UMEC/VI exercise studies [16], it may be that the ESWT lacks the sensitivity and repeatability in the multicentre setting to consistently detect benefits of UMEC/VI bronchodilator therapy on EET in patients with COPD for extended periods. This may be due to factors such as the operationalisation of the test, patient motivation, encouragement and learning effects, all of which are known to affect exercise testing but not accounted for in the ESWT [6, 25]. A recent trial investigated the effect of such interventions on EET in patients receiving tiotropium/olodaterol 5/5 µg [26]. Although every effort was made to reduce the impact of these factors on this study, it is impossible to ensure complete consistency across every ESWT as there are many nuances to the test (e.g. footwear, presence of a watch). The results of this trial should help further clarify the role of other factors, such as exercise training and behavioural modification, in exercise testing.
The MOVE study was a randomised, crossover study with two 21-day treatment periods, which assessed the effect of indacaterol/glycopyrronium 110/50 µg versus placebo on physical activity in patients with moderate-to-severe COPD [27]. Improvements in the activity-related energy expenditure were observed with indacaterol/glycopyrronium 110/50 µg compared with placebo after the first 21-day treatment period, but not after the second 21-day treatment period [27]. Although the MOVE study did not analyse EET, a similar pattern to that in the current study was observed, i.e. short-term benefits in exercise capacity were achieved with bronchodilator therapy. This highlights that further investigations are required to clarify the effect of long-acting muscarinic antagonist/long-acting β2-agonist combinations on exercise capacity.
A limitation of the current study was that measures of dynamic hyperinflation such as inspiratory capacity were not assessed during exercise testing. As such, it is not known whether UMEC/VI improved dynamic hyperinflation during exercise or if a loss of effect on dynamic hyperinflation may have contributed to the reduced EET response observed at week 12.
Conclusions
Treatment with UMEC/VI did not result in a significant improvement in ESWT at week 12 compared with placebo in patients with COPD, although improvements were observed at day 2 and week 6. Additionally, treatment with UMEC/VI provided sustained improvements over placebo in measures of lung function, hyperinflation and health status over 12 weeks.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary Materials 00073-2017_supplement
Disclosures
Supplementary Material
C.J. Kalberg 00073-2017_Kalberg
D.A. Lipson 00073-2017_Lipson
J.H. Riley 00073-2017_Riley
M. Shoaib 00073-2017_Shoaib
Acknowledgements
Editorial support (in the form of writing assistance, assembling tables and figures, collating author comments, grammatical editing, and referencing) was provided by Matthew Robinson (Fishawack Indicia, Abingdon, UK) and was funded by GSK.
Author contributions: J.H. Riley contributed to the study conception or design and data acquisition. C.J. Kalberg and A. Donald contributed to the study conception or design and data analysis or interpretation. D.A. Lipson, M. Shoaib and L. Tombs contributed to data analysis or interpretation. All authors were involved in the preparation and review of the manuscript, and approved the final version to be submitted.
Footnotes
This article has supplementary material available from openres.ersjournals.com
This study is registered at www.clinicaltrials.gov with identifier number NCT02275052.
Support statement: This study was funded by GlaxoSmithKline (GSK identifier 201317). Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at openres.ersjournals.com
- Received June 16, 2017.
- Accepted October 21, 2017.
- Copyright ©ERS 2018
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










