





Abstract
Viral infections predispose to the development of childhood asthma, a disease associated with increased lung mast cells (MCs). This study investigated whether viral lower respiratory tract infections (LRTIs) can already evoke a MC response during childhood.
Lung tissue from young children who died following LRTIs were processed for immunohistochemical identification of MCs. Children who died from nonrespiratory causes served as controls. MCs were examined in relation to sensitisation in infant mice exposed to allergen during influenza A infection.
Increased numbers of MCs were observed in the alveolar parenchyma of children infected with LRTIs (median (range) 12.5 (0–78) MCs per mm2) compared to controls (0.63 (0–4) MCs per mm2, p=0.0005). The alveolar MC expansion was associated with a higher proportion of CD34+ tryptase+ progenitors (controls: 0% (0–1%); LRTIs: 0.9% (0–3%) CD34+ MCs (p=0.01)) and an increased expression of the vascular cell adhesion molecule (VCAM)-1 (controls: 0.2 (0.07–0.3); LRTIs: 0.3 (0.02–2) VCAM-1 per mm2 (p=0.04)). Similarly, infant mice infected with H1N1 alone or together with house dust mite (HDM) developed an increase in alveolar MCs (saline: 0.4 (0.3–0.5); HDM: 0.6 (0.4–0.9); H1N1: 1.4 (0.4–2.0); HDM+H1N1: 2.2 (1.2–4.4) MCs per mm2 (p<0.0001)). Alveolar MCs continued to increase and remained significantly higher into adulthood when exposed to H1N1+HDM (day 36: 2.2 (1.2–4.4); day 57: 4.6 (1.6–15) MCs per mm2 (p=0.01)) but not when infected with H1N1 alone.
Our data demonstrate that distal viral infections in young children evoke a rapid accumulation of alveolar MCs. Apart from revealing a novel immune response to distal infections, our data may have important implications for the link between viral infections during early childhood and subsequent asthma development.
Abstract
Viral infections in children evokes a rapid recruitment and accumulation of mast cells in the alveolar parenchyma http://ow.ly/i9eN30meNM7
Introduction
Lower respiratory tract infections (LRTIs) are associated with a substantial disease burden in young children. Influenza A, respiratory syncytial virus (RSV) and adenovirus (ADV) are major causes of LRTIs including bronchiolitis, croup and pneumonia in infants [1]. Studies on the frequency of viral infections in developed countries have shown that infants and preschool children experience 6–10 viral infections annually [2]. The risk of developing asthma is increased with frequent viral-induced bronchiolitic episodes in combination with allergic sensitisation to airborne allergens [3, 4]. Albeit that there is strong evidence from longitudinal studies, it is not known how distal viral infections during early childhood are connected to the development of asthma [5, 6].
Previous studies have revealed a surprisingly high density of alveolar mast cells (MCs) in healthy adult lungs [7]. The presence of large numbers of MCs at mucosal surfaces such as the lung suggests that these cells may play a crucial role in the innate immune defence against pathogens, toxins and tissue damage [8]. Indeed, studies in animal models have shown increased mortality after infections in MC-deficient mice [9, 10]. In addition, increased numbers of bronchiolar MCs have been reported in an animal model of viral bronchiolitis and this increase was associated with hyperresponsiveness to methacholine [11]. However, almost nothing is known regarding the MC responses to viral infection in human lungs, especially in the alveolar parenchyma, where both human and animal data are lacking.
Although asthma has classically been considered a disease of the central airways, new data suggest that both bronchioles and the alveolar parenchyma are subjected to inflammation [12–18]. Interestingly, among the most marked alveolar changes in asthma is an expanded and activated pool of alveolar MCs [18, 19]. Whether a similar MC response occurs during LRTIs as well as the potential link between LRTIs and distal MC responses in asthma, remains unknown.
Here, we have studied the recruitment and heterogeneity of MCs in a unique lung tissue material from children who suffered from fatal LRTIs. Furthermore, the connection to allergic sensitisation and the development of an asthmatic phenotype was studied in a mouse model of influenza A infection and house dust mite (HDM) exposure in infant mice.
Material and methods
Model of influenza A infection and allergic inflammation in infant mice
15-day pregnant (female) BALB/c mice were purchased from Charles River Laboratories (Ottawa, ON, Canada), housed under specific pathogen-free conditions, and maintained on a 12-h light–dark cycle with food and water ad libitum. Upon birth, mothers were housed with their litters in light-protected cages until completion of the study (or weaning at 4 weeks of age). 8-day-old mice were infected with influenza A and, 7 days later, exposed to HDM 5 days a week for a total of 3 weeks. To investigate whether changes persisted, the protocol was recapitulated and mice were allowed to rest for a period of 3 weeks. Animals were monitored for signs of illness twice daily for a period of 10 days following infection. To evaluate the total number of MCs, 3-μm sections were prepared and the sections were stained with a 1% solution of toluidine blue. Thereafter, MCs per section were counted and related to tissue area using ImageScope (Aperio, Vista, CA, USA). All experiments described in this study were approved by the Animal Research Ethics Board of McMaster University (Hamilton, ON, Canada). For details, see the supplementary material.
Subjects
Lung samples from post mortem lung tissue from young children (median age (range): 4 (5–16) months) who died from fatal LRTIs was collected (RSV, n=5; ADV, n=10; and influenza A, n=6). Control lung tissues were obtained from 10 age-matched children (3 (0.4–24) months) who died from nonrespiratory causes. All tissue samples were collected with informed parental consent. This study was approved by the institutional medical ethical committee, Sao Paulo, Brazil, and at the Institute of Legal Medicine of the Medical School of the Hanover University, Hanover, Germany. For details, see the supplementary material.
Immunohistochemistry
Double immunohistochemical staining of MCTC and MCT
A double staining protocol was used for simultaneous visualisation of MCTC and MCT cells [7, 18, 20]. The staining was performed on paraformaldehyde-fixed, paraffin-embedded tissue by an automated immunohistochemistry robot. For details, see table 1 and the supplementary material.
Antibodies used for immunohistochemistry
Immunohistochemical identification of VCAM-1 and CD34, ITGB1/A4 and Ki-67+ MCs
Immunofluorescence double staining was used to visualise expression of CD34, integrin β1 (ITGB1), integrin α4 (ITGA4) or Ki-67 on tryptase-positive MCs. A single staining protocol was used for visualisation of VCAM-1. For details, see table 1 and the supplementary material.
Tissue analysis
Quantification of density of MC subtypes
High-resolution digital images of sections double-stained for MCTC and MCT were generated through a 20× microscope lens by an automated digital slide-scanning robot (Scanscope CS; Aperio). All MCs were quantified manually on blinded sections and related to the perimeter of the basal membrane (small airways and pulmonary vessels) or tissue area (alveolar parenchyma) using Visiomorph (Visiopharm, Hoersholm, Denmark) [18]. For details, see the supplementary material.
Quantification of VCAM-1 and CD34, ITGB1, ITGA4 and Ki-67+ MCs
The immunoreactivity per square millimetre of tissue area of VCAM-1 was calculated using Visiomorph. Tryptase-positive MCs and expression of CD34, ITGB1, ITGA4 and Ki-67 was analysed and the proportion (%) of positive MCs was calculated. For details, see the supplementary material.
Statistical analysis
Data were analysed using Kruskal–Wallis test with Dunn's multiple comparisons test for comparison among three groups or more and Mann–Whitney rank sum test was used for comparison between two groups using GraphPad Prism version 5 (GraphPad Software, Inc., La Jolla, CA, USA). The Spearman test (two-tailed) was used to study the correlations. Correlation analysis was made within pooled LRTI-infected children. Results were considered significant at p≤0.05.
Results
Increased number of MCs in a model of allergic inflammation during the course of an influenza A infection in infant mice
To address whether LRTIs directly contribute to increased pulmonary abundance of MCs in the context of allergic asthma, we investigated MCs in mice infected post-natally (8 days old) with influenza A (H1N1) and or challenged with HDM (figure 1a and b). The number of MCs, defined as purple toluidine blue-positive granulated cells [21], were quantified in the alveolar parenchyma of control mice and mice infected with influenza A, exposed to HDM, or both infected with influenza A and exposed to HDM, and normalised for tissue area. Only low numbers of MCs were detected in the lungs of saline- or HDM-treated infant mice (figure 1c, f and g). At day 36, MCs were increased in infant mice infected with influenza A and, even more so, in mice infected with influenza A with concomitant HDM exposure (figure 1a, b, h and i). When allowed further to recover for 3 weeks (day 57 of age), the MC numbers where only significantly increased in mice infected with influenza A and exposed to HDM (figure 1d and m). Higher levels of MCs were observed in mice infected with influenza A and exposed to HDM at day 57 compared to day 36 (figure 1e). No increase in MC numbers was observed in mice exposed to HDM alone at day 36 or 57 compared to control animals (figure 1c, d, g and k). At both time points, MC numbers were significantly increased in HDM+influenza A compared to HDM alone (figure 1c and d).
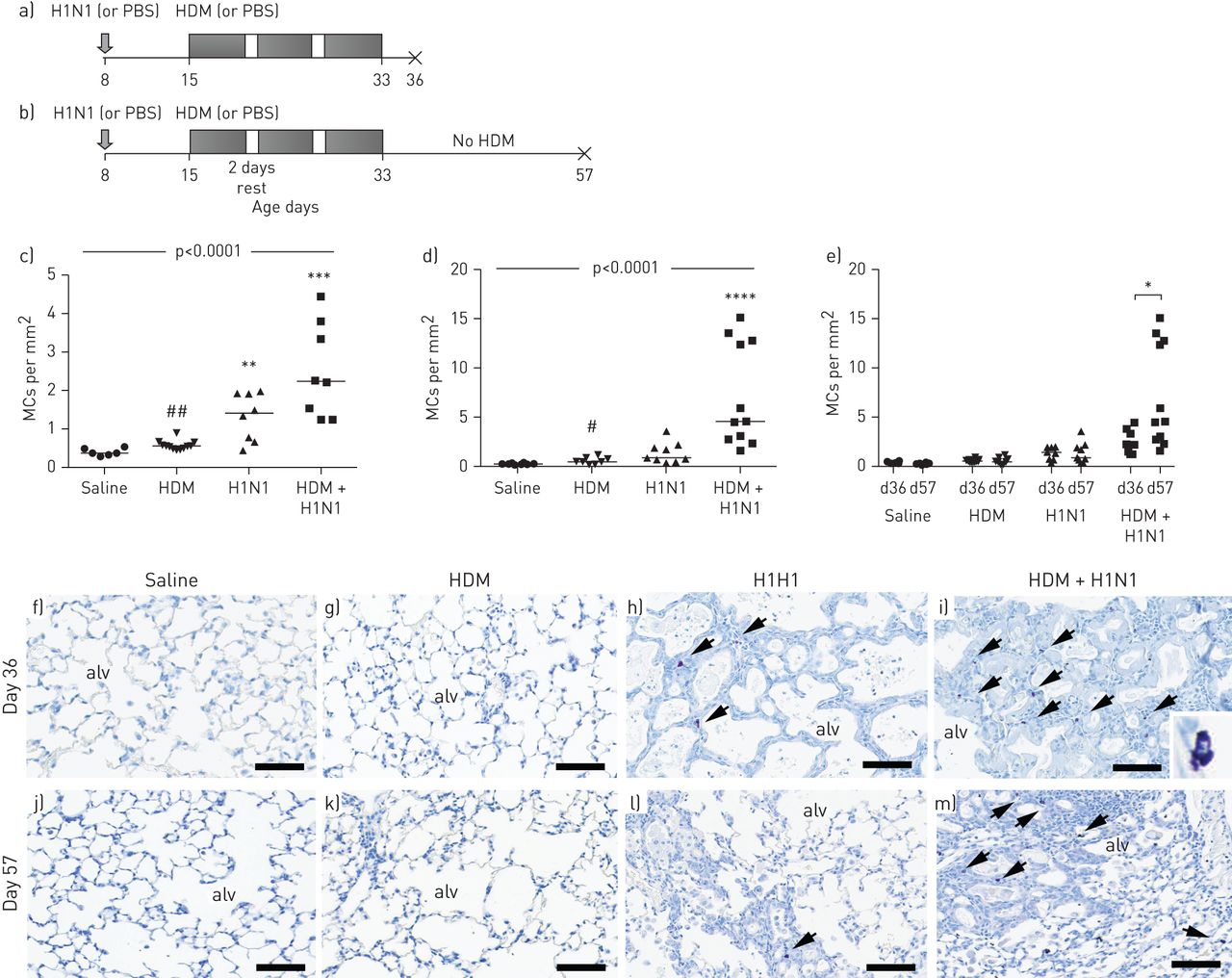
Experimental plan. a) 8-day-old mice were infected with influenza A virus (H1N1) or given PBS alone. 7 days later, PBS- and H1N1-infected groups were exposed either to 3 weeks of house dust mite (HDM) or saline and sacrificed 72 h after the last HDM exposure (day 36). b) Separate groups of 8-day-old mice were infected with influenza virus or given PBS alone. 7 days later, PBS- and H1N1-infected groups were exposed either to 3 weeks of HDM or saline, then, after the last HDM exposure, mice were rested for 3 weeks and sacrificed at 8 weeks of age (day 57). Mast cell (MC) densities in alveolar parenchyma in mice exposed to HDM or H1N1 alone or in combination compared to saline-treated animals: c) day 36 and d) day 57. e) MC densities at day 36 compared to day 57 in all groups. Toluidine blue stained slides (MCs shown in purple) picturing alveolar parenchyma from f and j) saline-, g and k) HDM-, h and l) H1N1-, and i and m) HDM+H1N1-treated animals. Scale bars=200 µm. Overall significance, using Kruskal–Wallis test, is denoted in each panel. Results were considered significant at p≤0.05, and individual differences between groups using Kruskal–Wallis test with Dunn's post hoc test. alv: alveolus. **: p≤0.01 versus controls; ***: p≤0.001 versus controls; ****: p≤0.0001 versus controls; #: p≤0.05 between HDM+H1N1 and HDM; ##: p≤0.01 between HDM+H1N1 and HDM.
Markedly elevated MC density in alveolar parenchyma in children who have died following LRTIs
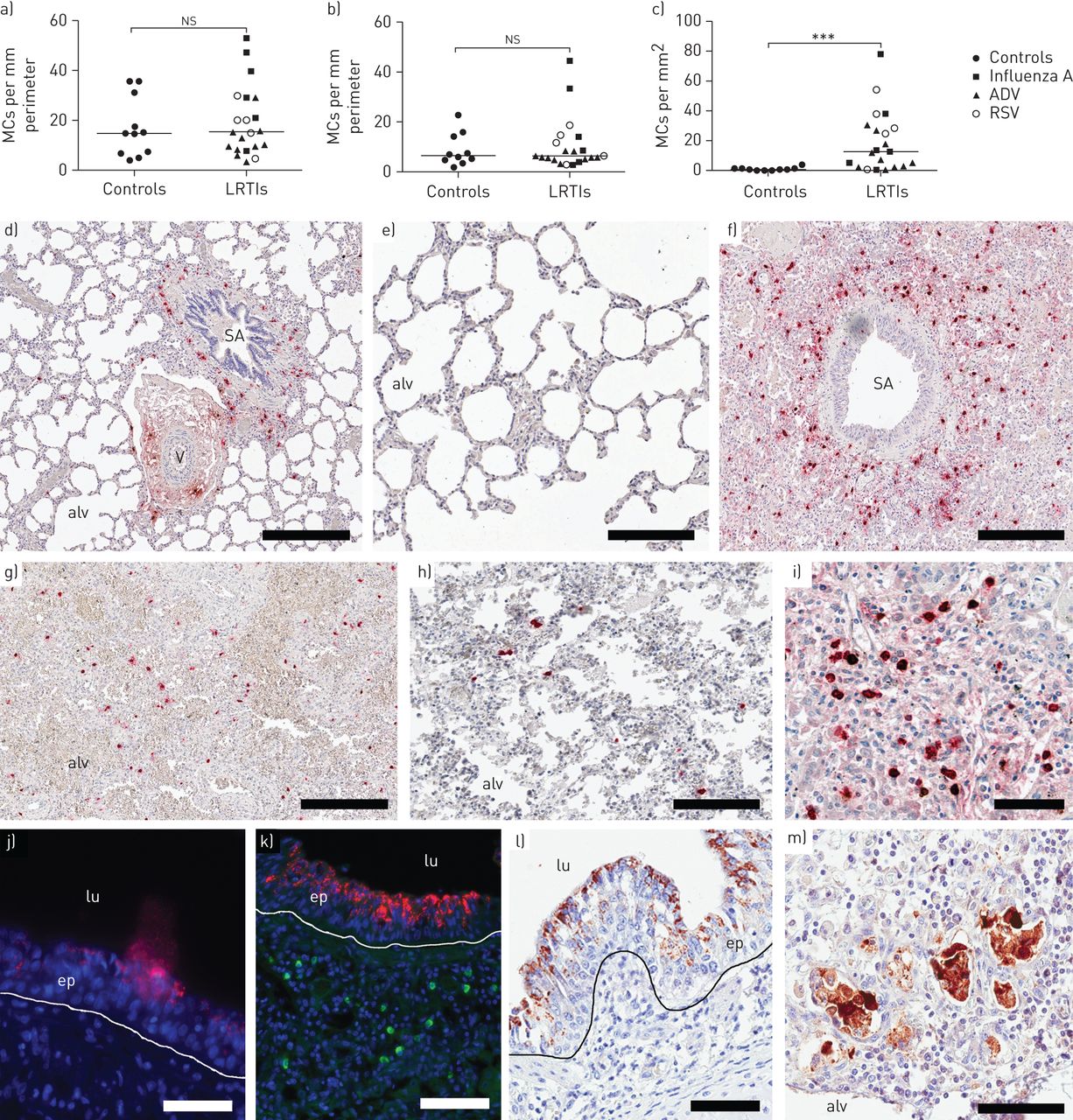
To further study the number of MCs under relevant human conditions, the density and anatomical localisation of distal lung MCs were quantified in the small airways (bronchioles), pulmonary vessels and the alveolar parenchyma in young children who died of LRTIs compared to controls. Interestingly, the control lungs virtually lacked alveolar mast cells (figure 2c and table 2), a result which contrasts the MC-rich alveolar parenchyma of adult healthy human lungs [7].

Mast cell (MC) densities in a) small airways, b) pulmonary vessels and c) alveolar parenchyma. Double immunohistochemical staining for tryptase (red) and chymase (brown). Representative photograph of d) a small airway and pulmonary vessel in a control lung from a child and e) alveolar parenchyma. f) Small airway in a lung from a child infected with influenza A. Alveolar parenchyma from young children who have died following g) respiratory syncytial virus (RSV), h) adenovirus (ADV) and i) influenza infections. Immunohistochemical staining for influenza A (red) in the epithelium with j and k) fluorescence and l) 3,3′-diaminobenzidine. k) Tryptase-positive MCs are shown in the subepithelium (green). m) RSV positivity in the alveolar parenchyma. Scale bars: d, f and g) 500 µm; e) 200 µm; h and k) 100 µm; i and m) 70 µm; j and l) 50 µm. Results were considered significant at p≤0.05. ns: not significant; LRTI: lower respiratory tract infection; SA: small airway; V: vessel; alv: alveolus; lu: lumen; ep: epithelium. ***: p≤0.001 using Mann–Whitney rank sum test, in comparison to controls.
Mast cell (MC) densities in young children with lower respiratory tract infections compared to controls
The density of MCs in small airways or the pulmonary vessels was not statistically altered in LRTI-affected lungs compared to controls (figure 2a, b, d, f, j and k). However, there was a marked and significant increase in MC numbers in the alveolar parenchyma in LRTIs (figure 2c, e, g–i and m). The increase was significant for both MCT and MCTC, subpopulations and present in RSV, ADV and influenza A infection. For details regarding different LRTIs and MC populations, see table 2.
Increased frequency of MC progenitors in the alveolar parenchyma after viral infections
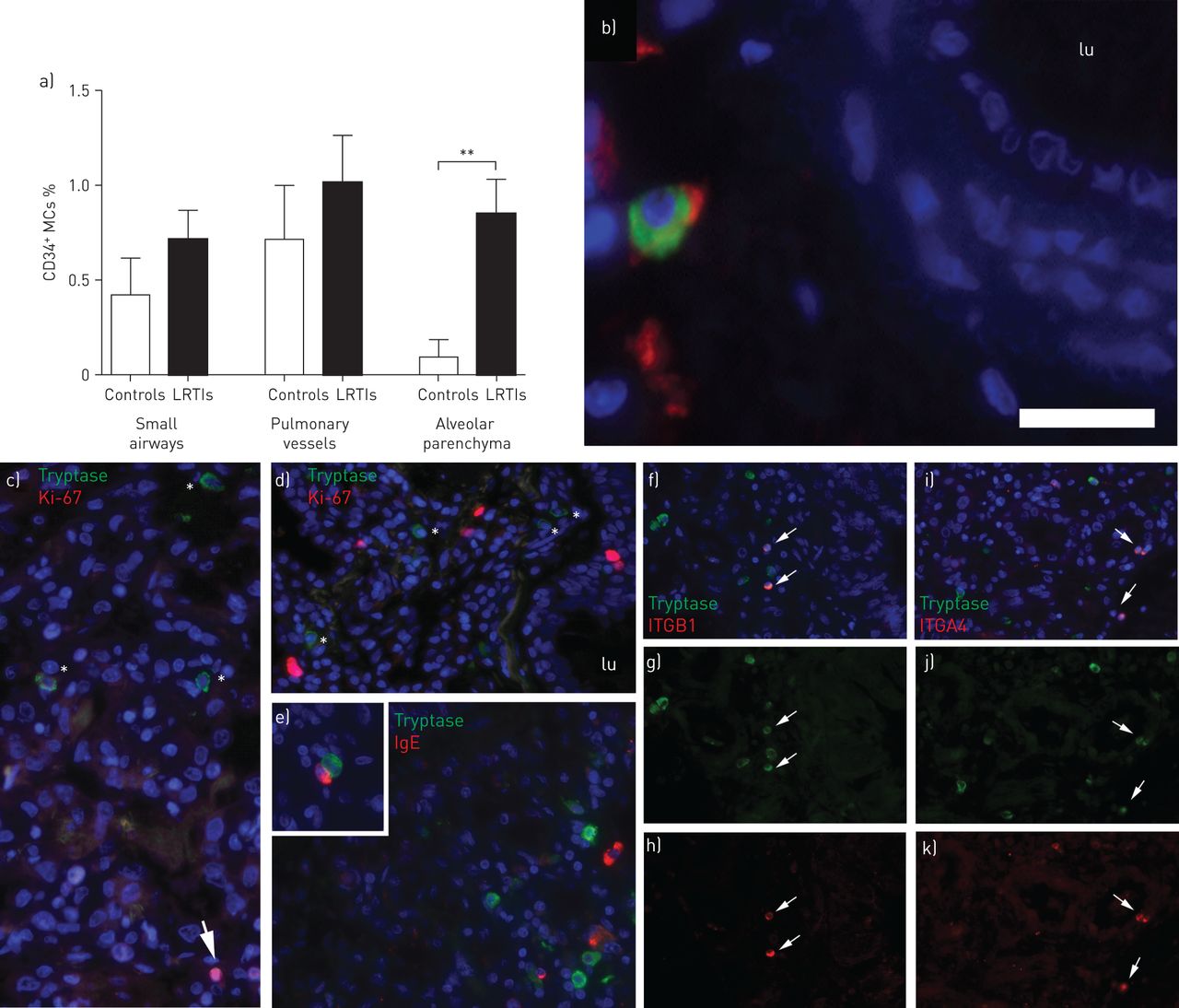
The median time with symptoms before death in the infected children were only 4 days (range 1–8 days). Since the LRTI-induced expansion of the alveolar MC pool thus took place rapidly after infection, we postulated that this would be linked with elevated levels of parenchymal MC progenitors, defined here as tryptase+ MCs double positive for the hematopoietic marker CD34. In small airways and pulmonary vessels, the frequency of MC progenitors was not significantly altered in children with LRTIs (figure 3a and b). The alveolar parenchyma, the only pulmonary structure where an accumulation of MCs was observed, contained a significantly higher frequency of MC progenitors (p<0.01) (figure 3a). Next, it was confirmed that mast cells in LRTI-affected lungs did express adhesion molecules integrin α4β1 that is thought to be critical for MC progenitor homing into tissue [22]. Although constituting a small proportion of all MCs, tryptase+ integrin α4β1+ MCs, phenotypically characterised as small and round, were clearly observed in the alveolar parenchyma of children who died following LRTIs (figure 3f–k).

a) Proportion of CD34+ mast cells (MCs) in small airways, pulmonary vessels and alveolar parenchyma. b) CD34+ (red) MCs (green) in a pulmonary vessel wall. Scale bar=30 µm). c and d) Influenza A-infected lung stained for tryptase (green) and proliferation marker Ki-67 (red). Arrows show Ki-67+ cells and * denotes tryptase+ cells. e) Alveolar parenchyma stained for tryptase (green) and IgE (red). Inset shows neighbouring IgE+ cell and MC. f–h) Alveolar parenchyma stained for tryptase (green) and integrin β1 (ITGB1) (red). i–k) Tryptase (green) integrin α4 (ITGA4) (red). Integrin+ MCs are denoted with arrows. Results were considered significant at p≤0.05. lu: lumen. **: p≤0.01 using Mann–Whitney rank sum test, in comparison to controls.
MCs do not proliferate locally in the lung of young children with LRTI
We next analysed the proliferation of MCs, defined as tryptase+ cells double positive for the proliferation marker Ki67. Scattered Ki-67+ cells were found throughout the lungs (figure 3c and d). Ki-67 immunoreactivity was not detected within tryptase+ cells, suggesting that the increased MC density was not dependent on local proliferation. In agreement with this, proliferative MCs were not found in adult lungs from patients with chronic obstructive pulmonary disease (COPD) or cystic fibrosis (data not shown). However, ∼2% of the total MCs (tryptase+) were identified as proliferating by Ki-67 immunostaining in bronchial lymph nodes from patients with very severe COPD (data not shown).
Our group has previously shown that FcεRI+ alveolar MCs and IgE bound to alveolar MCs is a hallmark of atopic asthma in adults [18, 19]. Immunofluorescence double staining revealed that lungs from both noninfected and LRTI-affected young children lacked MCs with surface-bound IgE (figure 3e). However, IgE+ CD138+ plasma cells were found in close proximity to MCs in the lungs of children with LRTI (inset in figure 3e).
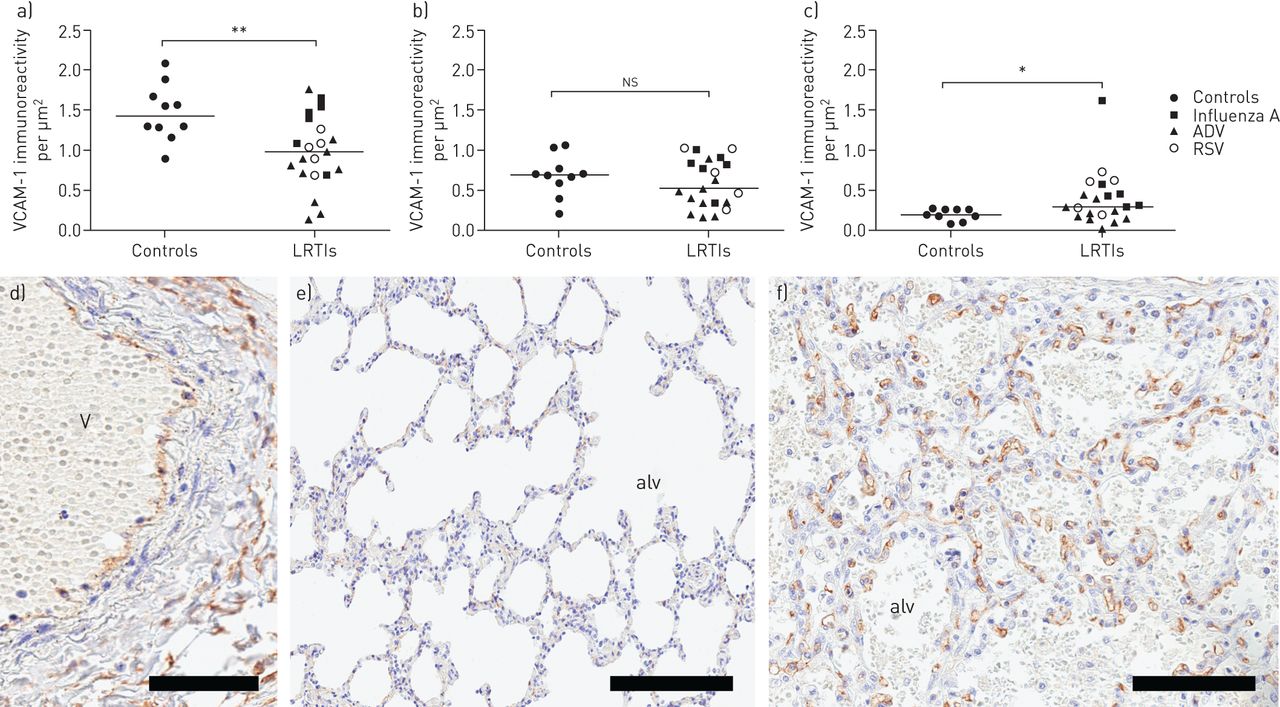
LRTI-affected lungs express elevated levels of the MC migration-facilitating molecule VCAM-1
Tissue recruitment and homing of MCs occurs through a process of endothelial VCAM-1 binding of MC integrins [23]. To investigate whether MC accumulation among children with fatal LRTI is associated with an induction of endothelial VCAM-1, the immunoreactivity of VCAM-1 was measured and related to sequential slides where the MCs were quantified. No difference in VCAM-1 expression was observed in small airway walls (figure 4a). In pulmonary vessels, the VCAM-1 density decreased compared to controls (figure 4b). In contrast, an increased VCAM-1 expression was observed in the alveolar parenchyma in LRTI-affected lungs compared to controls (figure 4c).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Vascular cell adhesion molecule (VCAM)-1 expression in a) small airway and b) pulmonary vessel walls, and c) alveolar parenchyma. d) Immunohistochemical staining of VCAM-1 in a pulmonary vessel from a child with lower respiratory tract infection (LRTI). VCAM-1 expression in alveolar parenchyma from e) a control child and f) a child with LRTI, respectively. Scale bars: d) 50 µm; e) 200 µm; f) 100 µm. Results were considered significant at p≤0.05. ns: not significant; ADV: adenovirus; RSV: respiratory syncytial virus; V: vessel; alv: alveolus. *: p≤0.05 using Mann–Whitney rank sum test, in comparison to controls; **: p≤0.01 using Mann–Whitney rank sum test, in comparison to controls.
Discussion
The results from our study demonstrate that a viral infection affecting the peripheral lung evokes a rapid accumulation of MCs in the alveolar parenchyma in both young children and mice. Apart from revealing a novel type of immune response in LRTI-affected human lungs, our experimental data suggest that virus-induced alveolar establishment of MCs in combination with allergen exposure in the distal lung may evoke a long-lasting immune alteration since MCs are long lived and are likely to remain in the tissue well after the infection has been cleared. Speculatively, LRTI-induced alveolar MC expansion early in life might therefore affect sensitisation and susceptibility towards allergens and asthma development later in life.
Many studies have suggested a connection between early-in-life viral infections, especially in the lower airways, and subsequent development of asthma. Children who are affected by viral LRTI during sensitisation by aeroallergens have up to a 30-fold increased risk of developing persistent or severe asthma, particularly when the events take place during postnatal lung growth [24]. The risk is affected by both intensity and frequency of the viral infection as well as allergen-specific IgE levels [25, 26]. The potential link between LRTI and asthma is further underscored by the fact that robust alveolar MC changes seem to be common features of adult asthma [18, 19, 27].
A main finding in our study is that a resident alveolar MC population was virtually absent in the nondiseased lungs from young children. This is in stark contrast with the healthy human adult lung where the highest density of MCs is found in the alveolar parenchyma [7]. Likewise, laboratory mice do not have resident alveolar MCs. In rodents, it is believed that such a population does not emerge with age unless subjected to a viral insult to the lung [28]. Indeed, our study shows that a viral infection evokes a rapid establishment of an alveolar MC population in mice and children. If this occurs during infancy, which is a critical stage of lung and immune response development, this could theoretically lead to increased susceptibility for sensitisation and asthma development later in life. Although our animal study showed an increase in MC numbers with influenza A alone, the numbers further increased with a combination of influenza A and HDM at both day 36 and 57. This might indicate that the inflammatory milieu caused by a combination of virus and allergen further enhances homing and/or survival of MC into the affected tissue. Although the methods used in our study are unable to evaluate MC activation, our analysis implicates that young children who suffered from LRTIs and survived, and who have susceptibility towards allergic sensitisation, may have a higher predisposition to react with an elevated MC response upon activation.
Due to their strategic position at mucosal surfaces MCs are among the first cell types to encounter foreign agents. As a consequence of this, MCs have been proposed to have important roles in the defence against pathogens [29–31]. Both in vivo and in vitro studies have demonstrated that MCs express several receptors that facilitate cellular responses to pathogens, including CD48, complement receptors, Fc receptors and Toll-like receptors [32–35]. In a recent study, Graham et al. [36] showed that cultured murine MCs are activated during influenza infection. Whereas activation and de novo production of cytokines is regulated by RIG-I/MAVS-dependent mechanisms, their study showed that degranulation is independent of this signalling pathway, indicating highly controlled MC activation during viral infections in mice. It was also shown that mice lacking MCs develop less severe inflammatory response and lung damage to influenza A compared to their wild-type counterparts [36]. Higher RSV-specific IgE titres have been shown in nasopharyngeal secretions of wheezing young children compared to young children who do not wheeze upon RSV infection [37–39]. Thus, virus-specific IgE might activate MCs and other FcεRI-expressing cells that results in the release of various pro-inflammatory mediators. Indeed, the nasopharyngeal concentration of the bronchoconstrictive MC mediator leukotriene C4 correlated with IgE titres in young children with RSV bronchiolitis [40]. Another evidence of infection-induced MC activation is that histamine levels are increased in patients during a virally induced asthma exacerbation [41]. An elevated MC response might therefore be part of the normal immune defence but the time point of infection, type of virus, or whether or not there is a simultaneous allergic sensitisation might predispose for asthma development. An expanded alveolar MC population in childhood, during a time-point of lung growth, is likely to have an impact on the development and remodelling of lung tissue as well as the immune responses and sensitisation towards harmful agents and allergens. Thus, establishment of an alveolar MC population early in life is likely to contribute to an aggravated inflammatory response against allergens or other pro-inflammatory agents that reach the distal lung [42, 43].
Migration of MCs to the airways has previously been shown in mouse models of allergic airway inflammation [44] and the increase in pulmonary MCs is preceded by a recruitment of MC progenitors from the blood into the lung [45]. For this recruitment to occur, MC progenitors need to express integrin α4β1 and α4β7, which interact with VCAM-1 on the lung endothelium [46]. In our study, we investigated whether the increase in lung MCs upon LRTI could result from recruitment of MC progenitors from the blood. We used CD34 in combination with tryptase to identify MC progenitors in the lung since tryptase is a MC specific marker and mature human MCs do not express CD34 [47, 48]. In young children infected with virus, this population of MC progenitors was increased within the alveolar parenchyma. It is likely that the frequency of MC progenitors is significantly underestimated using this combination of surface markers, as any MC progenitors that still have not developed tryptase-laden (i.e. CD34+ tryptase− cells) will not be identified with this staining panel. Although few in numbers, the MC progenitors were found to be significantly increased in the alveolar parenchyma of young children affected by viral infections, which together with the increased VCAM-1 expression may well have resulted in the higher total MC density in the peripheral lung. This is supported by animal studies of influenza A infection in mice [28]. Importantly, Brightling et al. [49] showed that the number of tryptase+ mast cells increased in the airway smooth muscle (ASM) in subjects with asthma compared with subjects with eosinophilic bronchitis and to that in normal controls. This group also showed that MCs within the ASM layer are linked to airway hyperresponsiveness [50]. Very few studies have investigated MCs in childhood asthma and some have found no difference in MC numbers in comparison to controls [51, 52]. An important study by Lezmi et al. [53] showed that the number of submucosal MCs was higher in symptomatic childhood asthma than in the paucisymptomatic group. They concluded that MCs are associated with severe exacerbations and submucosal eosinophilic inflammation in children with severe asthma, which suggests an important role for MCs in the development of asthmatic symptoms.
A study by Al-Garawi et al. [54] showed that infant mice are, unlike adult mice, hyporesponsive to HDM exposure. The hyporesponsiveness is, however, overcome in mice previously infected with influenza A infection. Furthermore, the pathological tissue changes persist into adulthood and are associated with reduced lung function. These findings correlate well to the alternations in MCs that we describe here. In the present study, we used tissue from the same experimental study [54] and found an increased number of alveolar MCs in influenza A and influenza A+HDM challenged mice, but not in mice exposed to HDM alone. The increase was significant in infected infant mice after 36 days but further elevated when combined with an allergic inflammation caused by HDM. The increase was still present after 57 days even though mice had been allowed to recover for 3 weeks and acute inflammation had subsided. At day 57, there were still patchy areas of remodelled lung architecture with MCs remaining in the influenza A+HDM mice. Our data indicate that the increased number of MCs persists well after the infection has cleared. This might give a predisposition to react to allergens, viruses and other MC stimuli in the future, and lead to asthma development later in life. However, it should be noted that the murine model used may not be equivalent to processes happening in the human lung, and that it is difficult to make this link without more human data. Although not in the scope of this study, it would be of great interest to explore the long-term presence of alveolar MCs even after the alveolar architecture has returned to the pre-infected state. Based on our result that only influenza A+HDM-treated mice had an increased MC population, one might speculate that the combination of HDM and influenza A causes the requirement for a maintained altered parenchymal architecture, which thereby supports MC survival and localisation.
In conclusion, by studying a unique human tissue material from young children who have died from LRTIs, we show here that distal viral infections cause a rapid de novo increase in MC populations in the alveolar parenchyma. The recruited MCs are likely to have important roles in the host defence against viruses. Our animal data further indicate that infection with influenza A and concomitant allergen exposure lead to abnormally elevated accumulation of MCs that persists into adulthood. Hence, LRTIs seem to represent an important mechanism of establishing a long-lasting presence of alveolar MCs, a population that is dramatically altered in adult asthma [12, 13]. Thus, children who suffer from a distal respiratory viral infection and survive are likely to have an increased resident alveolar MC population already in younger years, which speculatively affect their susceptibility to allergen sensitisation and development of asthma. Further research regarding the role of alveolar MCs in viral infection and the connection to asthma development under human in vivo conditions is warranted.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Acknowledgements
Special thanks to Anette Debertin and Thomas Tschernig (Hannover Medical School, Hannover, Germany) who performed the autopsies of the control children. We thank Karin Jansner and Britt-Marie Nilsson (Unit of Airway Inflammation, Lund University, Lund, Sweden) for skilful technical assistance with tissue processing and immunohistochemical staining. We also thank Göran Eriksson (Dept of Respiratory Medicine and Allergology, Lund University, Lund, Sweden) for reading and providing helpful comments on the manuscript.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Conflict of interest: C.K. Andersson has nothing to disclose.
Conflict of interest: M. Shikhagaie has nothing to disclose.
Conflict of interest: M. Mori has nothing to disclose.
Conflict of interest: A. Al-Garawi has nothing to disclose.
Conflict of interest: J.L. Reed has nothing to disclose.
Conflict of interest: A.A. Humbles is an employee of and holds shares in MedImmune LLC.
Conflict of interest: R. Welliver has nothing to disclose.
Conflict of interest: T. Mauad has nothing to disclose.
Conflict of interest: L. Bjermer has nothing to disclose.
Conflict of interest: M. Jordana has nothing to disclose.
Conflict of interest: J.S. Erjefält has nothing to disclose.
Support statement: This study was supported by the Heart and Lung Foundation, Sweden; the Swedish Medical Research Council; the Swedish Asthma and Allergy Association Research Foundation; and The Crafoord Foundation. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received March 7, 2018.
- Accepted October 5, 2018.
- The content of this work is copyright of the authors or their employers. Design and branding are copyright ©ERS 2018
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










