Article Text

Abstract
Background Pirfenidone is an oral antifibrotic agent that has been shown to reduce the decline in lung function in patients with idiopathic pulmonary fibrosis (IPF). We performed an integrated analysis of safety data from five clinical trials evaluating pirfenidone in patients with IPF.
Methods All patients treated with pirfenidone in the three multinational Phase 3 studies (CAPACITY (studies 004 and 006), ASCEND (study 016)) and two ongoing open-label studies (study 002 and study 012 (RECAP)) were included in the analysis. Safety outcomes were assessed during the period from the first dose until 28 days after the last dose of study drug.
Results A total of 1299 patients were included in the analysis. The cumulative total exposure to pirfenidone was 3160 person exposure years (PEY). The median duration of exposure was 1.7 years (range 1 week to 9.9 years), and the mean (±SD) daily dose was 2053.8 (±484.9) mg. Gastrointestinal events (nausea (37.6%), diarrhoea (28.1%), dyspepsia (18.4%), vomiting (15.9%)) and rash (25.0%) were the most common adverse events; these were generally mild to moderate in severity and without significant clinical consequence. Elevations in alanine aminotransferase or aspartate aminotransferase greater than three times the upper limit of normal occurred in 40/1299 (3.1%) patients (adjusted incidence, 2.3 per 100 PEY). Elevations were generally transient and reversible with dose modification or discontinuation.
Conclusions A comprehensive analysis of safety outcomes in a large and well-defined cohort of 1299 patients with IPF who were followed prospectively for up to 9.9 years demonstrated that long-term treatment with pirfenidone is safe and generally well tolerated.
Trial registration numbers NCT00287716, NCT00287729, NCT00662038, NCT01366209.
- Interstitial Fibrosis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
These findings represent a comprehensive analysis of safety outcomes in a large and well-defined cohort of 1299 patients with idiopathic pulmonary fibrosis treated with pirfenidone.
During this long-term, prospective follow-up of up to 9.9 years, pirfenidone was safe and generally well tolerated.
Gastrointestinal and skin-related events were among the most common adverse events and were generally mild-to-moderate in severity and responsive to dose modification.
Elevations of aminotransferases typically occurred within the first 6 months of treatment and were generally transient, reversible with dose modification or discontinuation and without clinical sequelae.
Background
Pirfenidone is a novel oral antifibrotic agent that is approved for the treatment of idiopathic pulmonary fibrosis (IPF)—a chronic, progressive, diffuse parenchymal lung disease of unknown aetiology characterised by irreversible declines in lung function and exercise capacity.1
The clinical efficacy and safety of pirfenidone in patients with IPF have been demonstrated in three multinational, randomised, double-blind, placebo-controlled, Phase 3 trials and one randomised, double-blind, placebo controlled, Phase 3 trial conducted in Japan.2–4 Clinical efficacy data from the multinational Phase 3 trials provided evidence of a meaningful treatment benefit on multiple measures of disease status, including forced vital capacity (FVC), 6-minute walking distance (6MWD) and progression-free survival. Gastrointestinal and skin-related events were the most commonly reported adverse events; these were generally mild to moderate in severity and rarely resulted in treatment discontinuation. In a pooled analysis of outcomes in the three multinational trials, there were fewer deaths due to any cause and fewer deaths related to IPF in the pirfenidone group compared with placebo.3
A subsequent integrated analysis of safety in 789 patients treated with pirfenidone in either of two Phase 3 studies (studies 004 and 006 (CAPACITY)) and/or one of two open-label studies (study 002 and study 012 (RECAP)) demonstrated that treatment with pirfenidone for up to 7.7 years was safe and generally well tolerated.5 Consistent with prior observations, gastrointestinal and skin-related events were common but were generally mild to moderate in severity and rarely led to treatment discontinuation. Elevations in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than three times the upper limit of normal (ULN) occurred in 2.7% of patients; however, all were transient, reversible with dose modification and without clinical sequelae.
To further evaluate the long-term safety and tolerability of pirfenidone, we performed a comprehensive analysis of safety outcomes in the integrated population from five clinical trials evaluating pirfenidone in patients with IPF, including three Phase 3 multinational trials and two long-term open-label studies. The present report includes data from an additional pivotal trial (ASCEND) and a substantially increased cumulative total duration of exposure to pirfenidone compared with the previously published integrated safety analysis.
Methods
Study subjects
All patients randomised to treatment with pirfenidone in the Phase 3 multinational studies (CAPACITY (studies 004 and 006), ASCEND (study 016)) and all patients receiving at least one dose of pirfenidone in either of two ongoing open-label studies (study 002 and study 012 (RECAP)) formed the integrated population. Study 002 is an open-label compassionate use study in the USA; study 012 is an open-label extension study evaluating pirfenidone in patients who completed any one of the three phase 3 multinational studies.
Eligibility criteria for the Phase 3 multinational studies have been previously described (see supplementary material, table S1).2 ,3 All patients who completed the final study visit in the Phase 3 multinational studies were eligible for enrolment in study 012. The original eligibility criteria for study 002 included a diagnosis of either IPF or secondary pulmonary fibrosis; secondary pulmonary fibrosis was eliminated as a diagnostic eligibility criterion in Protocol Amendment 1 (15 December 2003).
Study design
All patients who met the criteria for enrolment in the Phase 3 multinational studies were randomised to treatment with pirfenidone or placebo. In study 004, patients were randomly assigned (2:1:2) to one of three groups: pirfenidone 2403 mg/day, pirfenidone 1197 mg/day or placebo. In studies 006 and 016, patients were randomly assigned (1:1) to treatment with pirfenidone 2403 mg/day or placebo. Study drug was administered orally with food in three equally divided daily doses and escalated to the full dose during a 2-week dose titration period in all three studies. Patients were treated for a minimum of 72 weeks in studies 004 and 006 and for 52 weeks in study 016.
In study 012, all eligible patients were treated with oral pirfenidone 2403 mg/day administered according to the same schedule as in the Phase 3 studies. All patients were required to complete a 2-week dose escalation period, regardless of prior treatment assignment in the Phase 3 studies. In study 002, patients who enrolled prior to protocol amendment 2 (15 September 2005) received pirfenidone 40 mg/kg/day (maximum of 3600 mg/day) administered orally in three equally divided daily doses following a 2-week dose escalation period. Patients who enrolled after the protocol amendment were treated with a target maintenance dose of 2403 mg/day.
Physical examination and routine clinical laboratory tests were performed at prespecified intervals in all five studies. A directed medical history, including a review of adverse events, concomitant medications, hospitalisations and compliance with the assigned treatment regimen, was conducted at each study visit.
Statistical analysis
All analyses were conducted using data from the integrated population as of the interim data cut-off date of 17 January 2014. Demographic and baseline characteristics are summarised descriptively for the integrated population as well as for the pooled pirfenidone 2403 mg/day and placebo groups from the multinational Phase 3 trials.
The total duration of exposure to study drug is defined as the time between the first and last dose of study drug. For patients who were treated with pirfenidone in the Phase 3 studies and subsequently enrolled in the open-label extension study (study 012), total duration of exposure is based on the time from the first dose of study drug in the Phase 3 study to the last dose of study drug in the open-label extension study at the interim data cut-off date (17 January 2014). The cumulative total exposure is expressed as person exposure years (PEY); one PEY is equal to one patient exposed to study drug for 1 year. The mean daily dose of study drug was calculated over the full duration of use, excluding the 2-week titration period.
Treatment-emergent adverse events (TEAEs) are defined as adverse events with an initial onset or worsening following the first dose and within 28 days after the last dose of study drug. The crude and adjusted incidence rates of treatment-emergent death, defined as death due to any cause between the first dose and 28 days after the last dose of study drug, are summarised for the integrated population and by treatment assignment in the Phase 3 studies. The crude incidence was adjusted for differences in duration of exposure by using person-time in the denominator. The adjusted incidence of mortality is reported as deaths per 100 PEY (number of deaths divided by the total PEY for the group, multiplied by 100).
Analyses of liver-related outcomes included all patients who had at least 4 weeks of exposure to study drug. Crude and adjusted incidence rates are summarised for the integrated population and by treatment assignment in the Phase 3 studies. The crude incidence was calculated as the number of events divided by the total number of patients exposed to study drug. The adjusted incidence rate was calculated as the number of events divided by the total PEY for the group.
All study participants provided written informed consent, and the protocol for each study was approved by the ethics committee or institutional review board at each participating institution.
Results
The integrated population included a total of 1299 patients; of these, 710 (54.7%) were initially treated with pirfenidone in the Phase 3 studies (study 004, n=261; study 006, n=171; study 016, n=278), 506 (39.0%) were treated with placebo in the Phase 3 studies and initiated treatment with pirfenidone in study 012 and 83 (6.4%) initiated treatment with pirfenidone in study 002 (figure 1).

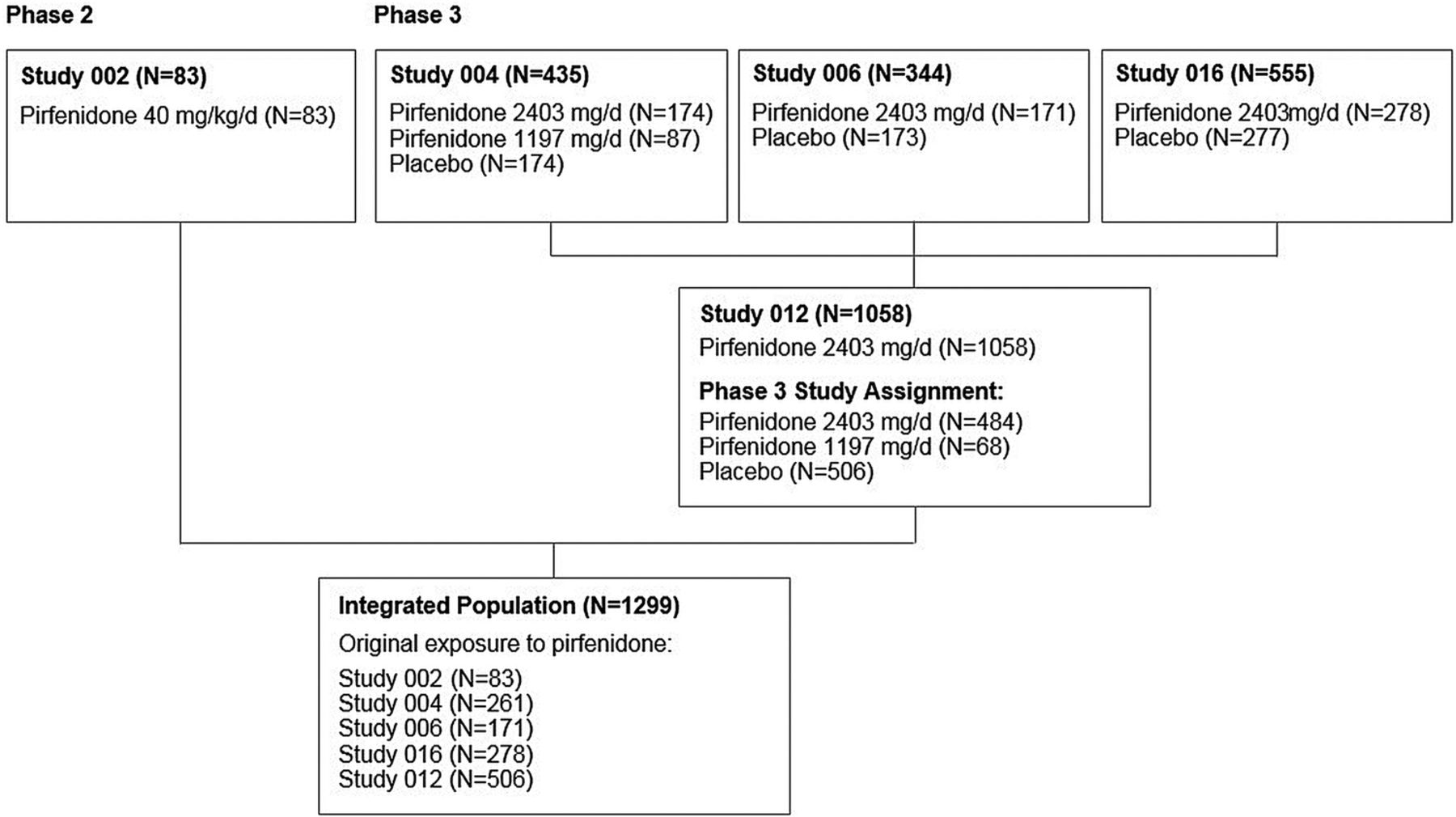{kind=link}
Study profile.
Demographics and baseline characteristics in the integrated patient population are summarised in table 1. The majority of patients were Caucasian (94.6%) and male (74.5%). A total of 97 (7.5%) and 210 (16.2%) patients in the integrated population had baseline values for FVC and carbon monoxide diffusing capacity (DLCO), respectively, that were below the minimum criteria for enrolment in the original Phase 3 CAPACITY studies, including 79 (6.1%) patients who had baseline DLCO values below 30%, the minimum criterion for enrolment in the ASCEND study (see online supplementary material, figure S1).
Demographics and baseline characteristics
Overall exposure to pirfenidone is summarised in table 2. The cumulative total exposure to pirfenidone was 3160 PEY. The median duration of exposure was 1.7 years (range 1 week to 9.9 years), and the mean (±SD) daily dose was 2053.8 (±484.9) mg. A total of 545 (42%) patients received pirfenidone for ≥2 years and 325 (25%) patients received pirfenidone for ≥4 years. The majority of patients (964 (74.2%)) received a mean daily dose between 1800 and 2600 mg.
Extent of exposure to pirfenidone
Consistent with prior observations in the pirfenidone and placebo groups in the Phase 3 clinical trials, almost all patients in the integrated population experienced at least one TEAE (see online supplementary material, table S2). A total of 495 (38.1%) patients in the integrated population discontinued treatment due to an adverse event, compared with 91 (14.6%) and 60 (9.6%) patients in the pooled pirfenidone and placebo groups in the Phase 3 trials. When adjusted for differences in duration of exposure, the rate of TEAEs in the integrated population was consistent with the rates in the Phase 3 trials. The most common adverse event leading to treatment discontinuation in the integrated population was IPF (11.5%); the only other events that led to discontinuation in >1% of patients were nausea (1.7%), rash (1.5%) and respiratory failure (1.3%).
The most common adverse events are presented in table 3. Gastrointestinal events (nausea (37.6%), diarrhoea (28.1%), dyspepsia (18.4%), vomiting (15.9%)) and rash (25.0%)) were among the most commonly reported adverse events in the integrated population; these were generally mild to moderate in severity and rarely led to treatment discontinuation (table 4). Additionally, the incidence of gastrointestinal and skin-related events was similar to the observed incidence among patients treated with pirfenidone in the Phase 3 trials, suggesting that the risk of such events does not increase with prolonged exposure. Respiratory adverse events, including cough, dyspnoea and IPF, were more common in the integrated population than in the pooled pirfenidone and placebo groups in the Phase 3 trials—a finding that is consistent with expectations in a population with a chronic progressive respiratory disease over a long duration of observation.
Treatment-emergent adverse events*
Gastrointestinal and skin-related adverse events of interest
Serious adverse events were reported by 639 (49.2%) patients in the integrated population, compared with 168 (27.0%) and 178 (28.5%) patients, respectively, in pooled pirfenidone and placebo groups in the Phase 3 trials. When adjusted for the longer duration of exposure, the rate of serious adverse events in the integrated population (49.8 per 100 PEY) was comparable to the observed rates in the pooled pirfenidone and placebo groups in the Phase 3 trials (41.7 and 44.2 per 100 PEY, respectively). The most common serious adverse events in the integrated population were worsening IPF (17.5%), pneumonia (7.9%), respiratory failure (3.2%), atrial fibrillation (2.8%) and bronchitis (2.7%). With the exception of atrial fibrillation, which occurred in 0.6% of patients in the pirfenidone and placebo groups in the Phase 3 trials, the incidence of each of these events was higher in the placebo group than in the pirfenidone group in the Phase 3 trials (see online supplementary material, table S3).
Analysis of liver chemistries in the integrated population showed no evidence of an increased risk of liver toxicity with longer durations of treatment. Elevations in ALT or AST greater than three times the ULN (>3×ULN) occurred in 3.1% of patients in the integrated population, compared with 3.7% and 0.8%, respectively, in the pooled pirfenidone and placebo groups in the Phase 3 trials (see online supplementary material, table S4). The adjusted incidence rate for ALT or AST elevations >3×ULN was 2.3 per 100 PEY in the integrated population, compared with 6.5 per 100 PEY in the pirfenidone group and 0.9 per 100 PEY in the placebo group during the Phase 3 trials. Elevations in aminotransferases were generally transient, reversible with dose modification and without significant clinical consequence. The Kaplan-Meier distribution for the time to onset of ALT or AST elevations >3×ULN showed that the majority of events occurred within the first 6 months of treatment (see online supplementary material, figure S2). Elevations in aminotransferases and/or serum bilirubin that occurred beyond 6–12 months of first exposure to pirfenidone could typically be attributed to other causes. Liver-related serious adverse events occurred in 1.0% of patients in the integrated population, compared with 1.0% and 0.2%, respectively, in the pooled pirfenidone and placebo groups in the Phase 3 trials.
A total of 233 (17.9%) deaths occurred between the first dose and 28 days after the last dose of study treatment in the integrated population (adjusted incidence, 7.4 per 100 PEY). The adjusted incidence of treatment-emergent death among the pooled treatment groups during the Phase 3 trials was 3.7 per 100 PEY in the pirfenidone group and 5.9 per PEY in the placebo group.
Discussion
We performed a comprehensive analysis of safety outcomes in a large and well-defined cohort of patients with IPF who were treated with pirfenidone and followed prospectively for up to 9.9 years (median 1.7 years; IQR 0.7–4.0 years). Our results provide further evidence that long-term treatment with pirfenidone is safe and generally well tolerated. Consistent with previous reports,2–9 gastrointestinal and skin-related events were among the most common adverse events; these were generally mild to moderate in severity and responsive to dose modification or other adverse event mitigation strategies. Of note, study subjects in the Phase 3 trials and the open-label extension study were instructed to take pirfenidone with food and advised to avoid sun exposure and to use sun block during treatment. Additionally, protocol-defined dose modification guidelines for gastrointestinal and skin-related adverse events were employed in the Phase 3 and open-label extension studies and subsequently incorporated into the product label.10 ,11 The generally transient nature of these events and the relatively low incidence of treatment discontinuation due to gastrointestinal or skin-related events over a longer duration of exposure provide further support for these recommendations.
The crude incidence of serious adverse events (unadjusted for differences in duration of exposure) was higher in the integrated population compared with that in the pooled pirfenidone and placebo groups in the Phase 3 clinical trials. However, this difference was driven largely by the increased incidence of worsening IPF—an expected finding given the irreversible nature of the disease and the long duration of observation.
Clinically significant elevations in aminotransferases occurred in a lower proportion of patients in the integrated population than in the pooled pirfenidone group in the Phase 3 trials. Analysis of serial laboratory results showed that these tended to occur within the first 6 months of treatment and were generally transient, reversible and without clinical sequelae. However, in light of the higher incidence of aminotransferase elevations in the pirfenidone group compared with placebo in the Phase 3 trials, the recommendation in the product labels to perform liver chemistry tests at monthly intervals for the first 6 months of treatment and at 3-month intervals thereafter remains consistent with prudent medical management.10 ,11
The strengths of our study include the large and well-defined study population the rigorous, prospective collection of longitudinal safety data and the duration of exposure to study drug of up to 10 years. To the best of our knowledge, the cumulative total exposure of 3160 PEY represents the largest and longest experience to date with any specific therapy in patients with IPF. Additionally, the integrated population comprised patients with a broad range of physiological impairment, including those with baseline values for FVC and DLCO that mandated exclusion from the Phase 3 clinical trials. The study population is therefore broadly reflective of the general population of patients with IPF, thereby enhancing the generalisability of the findings.
The findings of our study should be interpreted in the context of certain limitations. First, two of the five studies included in the analysis were open-label studies that did not include a parallel control group. Second, while study eligibility was generally broad, patients with certain comorbidities such as renal failure requiring dialysis and severe hepatic impairment or those receiving selected concomitant medications were excluded from enrolment in the studies included in our analysis (see online supplementary material, appendix 1). The extent to which the findings are generalisable to patients with these and other excluded comorbidities or to patients taking medications that were prohibited during the clinical trials, is therefore not known. Finally, clinical efficacy outcomes, such as 6MWD, DLCO and dyspnoea, were not assessed in the open-label studies. While serial measures of FVC were performed in these studies, the absence of a control group precludes meaningful interpretation of the results. Due consideration should therefore be given to the limited availability of efficacy data beyond 72 weeks in the assessment of the relative benefits and risks associated with long-term pirfenidone treatment.
Conclusion
A comprehensive analysis of safety outcomes in a large and well-defined population of 1299 patients with IPF who received pirfenidone for up to 9.9 years demonstrated that long-term treatment with pirfenidone is safe and generally well tolerated.
Acknowledgments
The authors wish to thank Kenneth Glasscock for medical writing and editorial assistance and also thank the participating medical staff and patients at all the study centres.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors CA, WZB, UC, RMdB, TEK, PWN and DV participated in the conception, design and execution of the CAPACITY study and in the acquisition and interpretation of original data. WZB, IG, MKG, EAF, RSF, TEK, LL, DJL, ZL, SDN, CAP, JJS and PWN participated in the conception, design and execution of the ASCEND study and in the acquisition and interpretation of original data. CA, WZB, UC, RMdB, TEK, PWN and DV participated in the conception, design and execution of the RECAP study, and all the authors participated in the interpretation of data from RECAP. ZL performed the statistical analyses for all data presented in the report. WZB and LL participated in the preparation of the initial draft of the manuscript. CA, UC, RMdB, IG, MKG, EAF, RSF, TEK, DJL, ZL, SDN, CAP, JJS and DV participated in the review and critical revision of the manuscript for intellectual content. All the authors approved the final draft and vouch for the accuracy of the overall content of the manuscript.
Funding This study was sponsored by InterMune Inc (Brisbane, CA).
Competing interests LL, IG, MKG, EAF, DJL, TEK, SDN, CAP, JJS and PWN were members of the ASCEND study steering committee. CA, UC, RMdB, TEK, PWN and DV were members of the CAPACITY study steering committee. LL has served on a scientific advisory board for Boehringer Ingelheim and InterMune and as a consultant to Boehringer Ingelheim. IG has received honoraria from Boehringer Ingelheim. CA has served on a scientific advisory board for InterMune. UC has served as a member of a study adjudication committee for Gilead and as a consultant for Bayer, Boehringer Ingelheim, Centocor and Roche. RMdB has served on a scientific advisory board for Actelion, Boehringer Ingelheim, InterMune and Novartis. DJL has served on a scientific advisory board for Boehringer-Ingelheim, Gilead, Immune Works and InterMune. TEK has served as a consultant for Actelion, Boehringer Ingelheim, Daiichi Sankyo, Immune Works and InterMune. SDN has served on a scientific advisory board and received research funding from Intermune; he has also received research funding and served as a consultant for Boehringer Ingelheim, Gilead and Roche/Genentech. CAP has received a research grant from InterMune. JJS has served on a scientific advisory board and received research funding from InterMune, and served as a consultant to Boehringer Ingelheim and Roche. PWN has served as a consultant for Boehringer Ingelheim, Bristol-Myers Squibb, InterMune, Moerae Matrix, Roche and Takeda. DV has served on a scientific advisory board for InterMune and received travel grants from Boehringer Ingelheim. WZB, EAF, RSF and ZL are employees of InterMune.
Ethics approval Food and Drug Administration.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.
Linked Articles
- Editorial