





Abstract
Chronic airway infections determine most morbidity in people with cystic fibrosis (CF). Herein, we present unbiased quantitative data about the frequency and abundance of DNA viruses, archaea, bacteria, moulds and fungi in CF lower airways.
Induced sputa were collected on several occasions from children, adolescents and adults with CF. Deep sputum metagenome sequencing identified, on average, approximately 10 DNA viruses or fungi and several hundred bacterial taxa.
The metagenome of a CF patient was typically found to be made up of an individual signature of multiple, lowly abundant species superimposed by few disease-associated pathogens, such as Pseudomonas aeruginosa and Staphylococcus aureus, as major components. The host-associated signatures ranged from inconspicuous polymicrobial communities in healthy subjects to low-complexity microbiomes dominated by the typical CF pathogens in patients with advanced lung disease. The DNA virus community in CF lungs mainly consisted of phages and occasionally of human pathogens, such as adeno- and herpesviruses. The S. aureus and P. aeruginosa populations were composed of one major and numerous minor clone types.
The rare clones constitute a low copy genetic resource that could rapidly expand as a response to habitat alterations, such as antimicrobial chemotherapy or invasion of novel microbes.
Abstract
The CF lung metagenome is composed of few viruses and fungi and hundreds of bacterial species, clones and subclones http://ow.ly/ZiqUE
Lessons for clinicians
More than 1000 microbial species were identified in the airways of children, adolescents and adults with cystic fibrosis (CF) by high-throughput shotgun metagenome sequencing of induced sputum samples.
Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria and Proteobacteria constitute >99% of the bacterial community in CF airways.
The DNA virome mainly consists of bacteriophages and occasionally of human pathogens, such as adeno- or herpes viruses and the mycobiome mainly consists of Candida and Aspergillus species.
A normal microbial airway metagenome lacking any CF-associated pathogens was observed in healthy CF adults with normal spirometry and normal lung clearance index. These index cases have been regularly seen since the age of diagnosis suggesting that continuous surveillance and intervention, if indicated, could prevent or decelerate progressive CF lung disease in some, but not all, subjects.
Pseudomonas aeruginosa was present in all samples from exocrine pancreatic insufficient (PI) CF patients, albeit prevalence was very low in subjects classified as P. aeruginosa-negative by standard bacteriology. P. aeruginosa is apparently acquired early in life in all PI CF patients. This novel information should be considered in forthcoming best practise guidelines for infection control and P. aeruginosa eradication programmes.
At present, the clonal populations of the common CF pathogens Staphylococcus aureus and P. aeruginosa in CF airways are more complex than presumed. Besides one or two major clone types that are amenable to standard culture-dependent diagnostics, several rare clone types coexist in CF lungs which could rapidly expand as a response to antimicrobial chemotherapy or invasion of novel microbes.
Introduction
CF is a life-shortening, debilitating, autosomal recessive disease that is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1]. The basic defect of impaired epithelial chloride and bicarbonate secretion predisposes CF patients to chronic airway infections with opportunistic pathogens, which determine most morbidity in people with CF [1]. Epidemiological data drawn from culture-dependent diagnostics of respiratory specimens indicated that the airways of CF patients become colonised with Haemophilus influenzae and Staphylococcus aureus during early childhood, followed later in life by Pseudomonas aeruginosa and sometimes by organisms such as Burkholderia cepacia complex or atypical mycobacteria [2]. Culture-independent technologies, however, revealed that the CF respiratory tract is not inhabited by these few pathogens, but rather by complex polymicrobial communities [3–10]. Sequencing of PCR-amplified parts of bacterial 16S ribosomal (r)DNA genes could identify over 100 distinct genera including Streptococcus and numerous anaerobes as major players that are routinely not detected during the culture-dependent processing of CF-derived respiratory secretions.
Sequence variations in the ancient and ubiquitous ribosomal RNA genes are considered to reflect the universal molecular clock of life [11]. Correspondingly, the composition of microbial communities is described by the abundance of individual rDNA sequences. However, one has to accept some inherent biases of this approach. First, prokaryotes and eukaryotes need to be analysed separately due to their basic difference of rDNA sequence and, correspondingly, most work has been confined to the bacterial microbiota. Secondly, the sequences of the sample are prepared for analysis by oligonucleotide primer-based amplification steps. Subsequent sequencing of the amplicons has a limited ability to resolve taxonomic identification to the species level, may fail to detect phyla and can skew the estimation of species relative abundance in a community [12, 13].
These limitations can be overcome by whole-genome shotgun sequencing [14], which may provide information about the composition of the community up to the level of clonal complexes. Within the context of the CF lungs, the published studies to date have focused on the sputum metagenomes derived from CF adults with advanced lung disease [15–17]. Each of the 10 so far investigated subjects hosted a unique polymicrobial community.
This study extends the scope to patients of all age groups and grades of disease severity in order to cover the whole range of lower airways microbial metagenomics in CF. Induced sputa collected from exocrine pancreatic sufficient (PS) and PI children, adolescents and adults with CF were investigated by whole-genome shotgun sequencing in order to identify the bacteria, archaea, DNA viruses, moulds and fungi residing in the respiratory secretions. Normalisation provided quantitative data about the relative and absolute abundance of microbial species. Moreover, by focusing on the major CF pathogens S. aureus and P. aeruginosa the metagenome sequence data sets were examined for the number of coexisting clone types and uncommon, probably de novo, mutations in antimicrobial resistance determinants. In future, such a metagenome-guided deep insight into clone and sequence variations could assist in our management of respiratory tract infections.
Methods
Patients
Subjects with CF were recruited from the CF clinic of Hanover Medical School (Hannover, Germany). All patients had been regularly seen at the CF clinic since the age of diagnosis. The diagnosis of CF had been made by the detection of two disease-causing mutations in the CFTR gene [18], and elevated chloride concentrations in the Gibson–Cooke pilocarpine iontophoresis sweat test [19] or a Sermet score in the CF range of nasal transepithelial potential difference measurements [20, 21] and/or chloride secretory responses in the CF range of intestinal current measurements [22]. Exocrine pancreatic status was assessed by the fecal elastase-1 test [23]. Lung function was assessed by spirometry, body plethysmography and, in the healthier subjects, multiple breath nitrogen washout [24, 25]. The 19 exocrine PI patients were homozygous for the most common CF mutation, p.Phe508del [18]. The 11 PS subjects were either compound heterozygous for a PI- and a PS-conferring CFTR mutation (n=10) [18] or homozygous for a PS mutation (n=1) [18]. At the date of recruitment patients were either 8–13 years old (children, group A), 18–23 years old (adolescents and young adults, group B) or >28 years old (adults, group C) (table 1). Patients were also classified by disease severity. Healthy CF subjects had normal anthropometry (body mass index >19 kg·m−2) and normal lung function, i.e. multiple breath nitrogen washout revealed a normal lung clearance index and spirometry yielded forced expiratory volume in 1 s (FEV1) values >90% predicted. Mildly affected CF patients (category “mild”) exhibited normal anthropometry, an anomalous lung clearance index and FEV1 values of 70–110% pred on the day of recruitment. Lung function was chronically compromised for ≥3 years in all moderately or severely affected CF patients (FEV1 50–70% pred, category “moderate”; FEV1 30–50% pred in the absence of an acute pulmonary exacerbation, category “severe”; FEV1 <30% pred in the absence of an acute pulmonary exacerbation, category “end-stage lung disease”). The clinical status of the individual subjects is described in table S1. The study was approved by the Ethics Committee of Hannover Medical School (no. 1510-2012).
Study participants
Wet lab experimental procedures
The procedures are described in the supplementary material. Briefly, induced sputum was collected by autogenic drainage during cycles of 3-min inhalation of 3% (w/v) hypertonic saline. After sputa had been diluted with buffer and subjected to hypotonic lysis, DNA was purified from the suspension according to the Hard-to-lyse-Bacteria protocol with the NucleoSpin Tissue kit (Machery-Nagel, Düren, Germany). DNA libraries were prepared from sheared DNA according to an in-house protocol. Sequencing was performed on a SOLiD 5500XL system (Life Technologies, Carlsbad, CA, USA) in colour space with 75 bp read length and implemented Exact call chemistry (Life Technologies).
In silico analyses
Approaches and software are described in the supplementary material. In brief, the raw sequence reads were trimmed and then (in this order) low quality reads, human reads, non-human low-complexity reads and non-human reads encoding mobile genetic elements were removed. The remaining microbial reads were normalised by GC content and genome length. This curated data set was then used for the identification of taxa (DNA viruses, bacteria, archaea, moulds and fungi), principal component analysis, search for mutation in antimicrobial resistance genes and analysis of the S. aureus and P. aeruginosa populations in the respiratory secretions.
Results
Lower airways microbial metagenome of PS and PI individuals with CF
Induced sputa were collected within 1 year in 6- to 20-week intervals from 11 PS (n=1–4, median two samples) and 19 p.Phe508del homozygous PI subjects with CF (n=1–5, median two samples). Deep metagenome sequencing identified, on average, <10 DNA viruses or fungi and several hundred bacterial taxa in samples from children (group A), adolescents (group B) and adults (group C) (figure 1b, figure S1, tables S2 and S3). Bacteria typically made up > 99% of the microbial community (figure 1a) [26]. The median contribution of DNA viruses and fungi was on average <1%, but it varied between 0.002% and 11% in the individual sample. The dominant taxa in the cumulative metagenome of the whole cohort were the bacterial species that are most frequently reported from culture-dependent diagnostics, i.e. streptococci, staphylococci, pseudomonads, Haemophilus sp., Burkholderia sp. and Stenotrophomonas sp. (figure 1c, table S3). Thus, quantitative metagenomics fits with culture-based epidemiological data for the most abundant bacterial species in CF lungs.
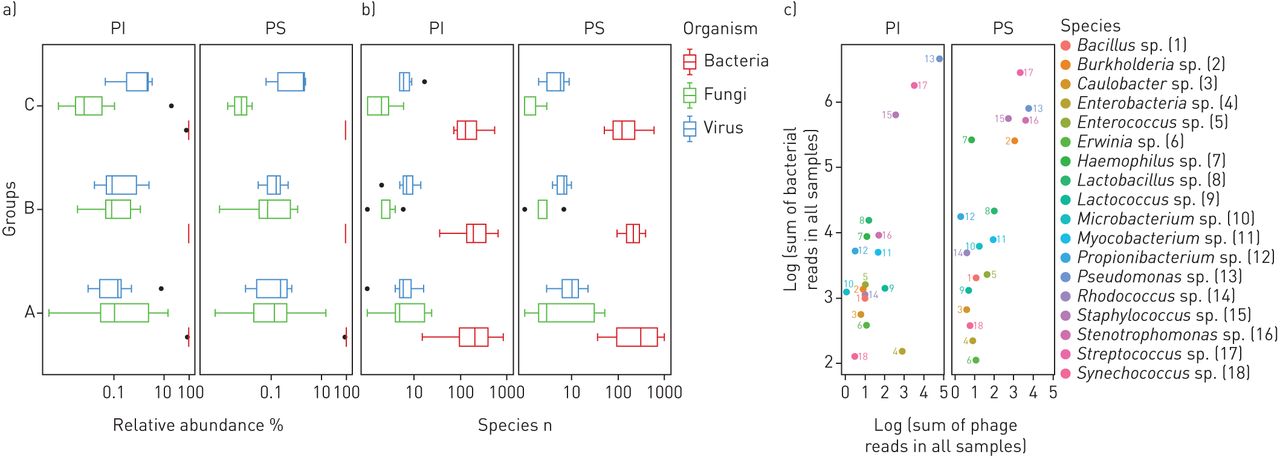
The cystic fibrosis (CF) lower airways metagenome: cumulative presentation of data sets. a, b) Box plot presentation of the absolute frequency and the relative abundance of DNA viruses, bacteria and fungi identified in induced sputa sampled from the lower airways of exocrine pancreas insufficient (PI) and exocrine pancreas sufficient (PS) children (A), adolescents (B) and adults (C) with CF. c) The dots in the double logarithmic plot depict the sum of reads identified for bacteriophages and their corresponding bacterial hosts at the genus level. Genera are differentiated by number and colour.
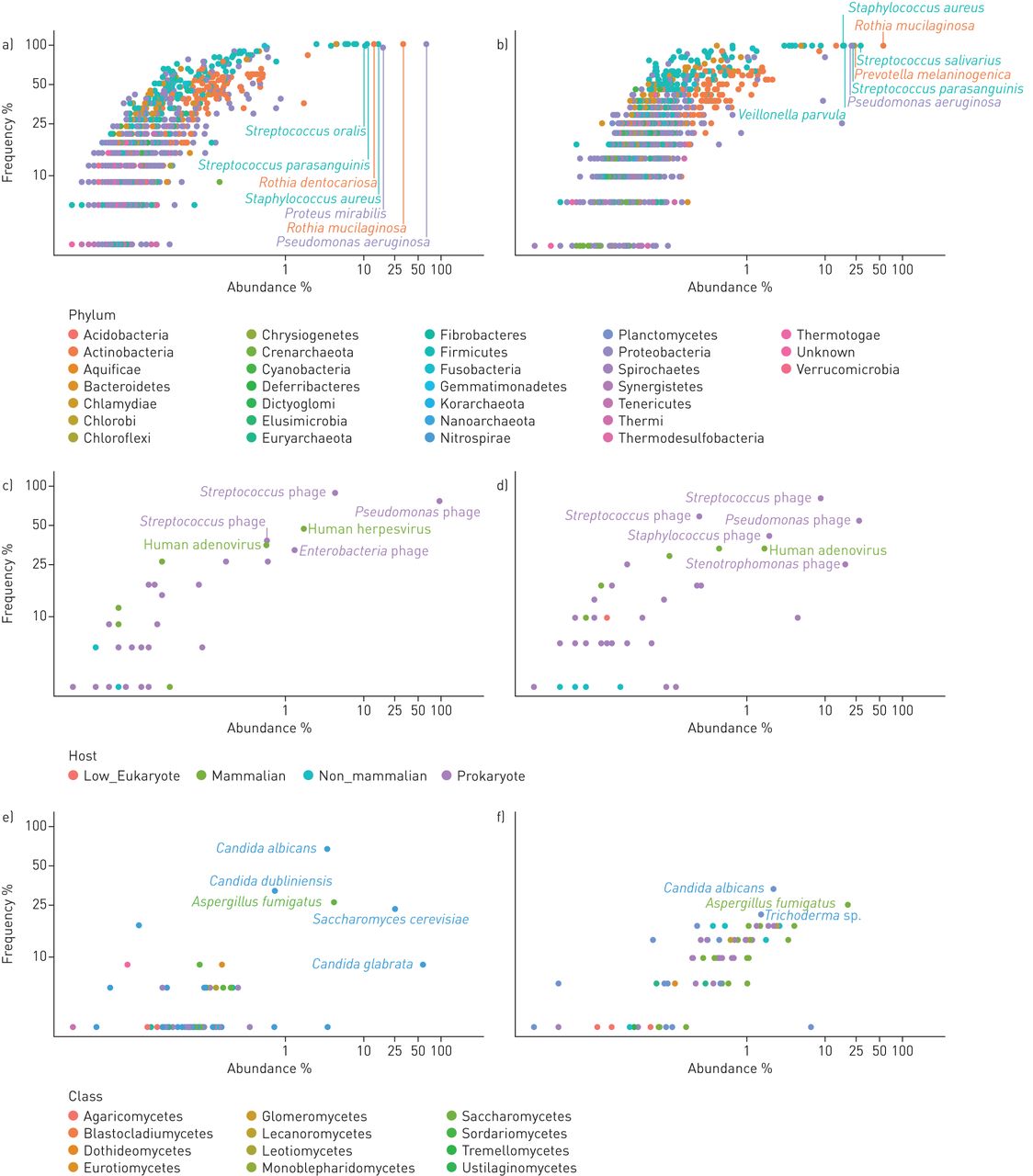
Figure 2 provides a detailed overview of the frequency of recovery of all detected species and their relative abundance. Taxa and primary data are listed in tables S1 and S3. The viral community consisted primarily of phages (figure 1c), a few human pathogens, primarily herpes virus and adenovirus, and rare cases of viruses infecting non-mammalian eukaryotic hosts. Dominant species in the mycobiome were Aspergillus species and Saccharomycetes including Candida sp. which had an average abundance of 4.3% and 94.5% in the mycobiome, respectively, consistent with current knowledge of CF mycology [27]. The community of bacteria and archaea turned out to be highly diverse, including numerous very lowly abundant species and phyla that have not yet been reported to inhabit the niche of the human CF lung. In total 1049 and 977 species belonging to 30 phyla were identified in the sputa from PS and PI patients, respectively (table S4). All archaeal phyla and 21 of the 26 bacterial phyla each had an abundance of <0.1%. The remaining five phyla, i.e. Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria and Proteobacteria, made up 99.8% and 99.9% of the bacterial community in PS and PI CF sputa, respectively (table 2). Besides the common CF pathogens S. aureus, P. aeruginosa, H. influenzae and Burkholderia spp., species belonging to the genera Rothia, Proteus and Streptococcus were most common among aerobes and facultative anaerobes. The community of anaerobic bacteria consisted, on average, of 98% of Prevotella melaninogenica and Veillonella parvula (table S4), typical inhabitants of the oral cavity that were also prominent members in the airway metagenome of healthy controls (table S5). Moreover, four further anaerobes were regularly identified as minor members in CF sputum, i.e. the oral bacteria Atopobium parvulum and Fusobacterium nucleatum, the intestinal inhabitant Megasphaera elsdenii and the zoonotic airway pathogen Streptobacillus moniliformis (table S4).
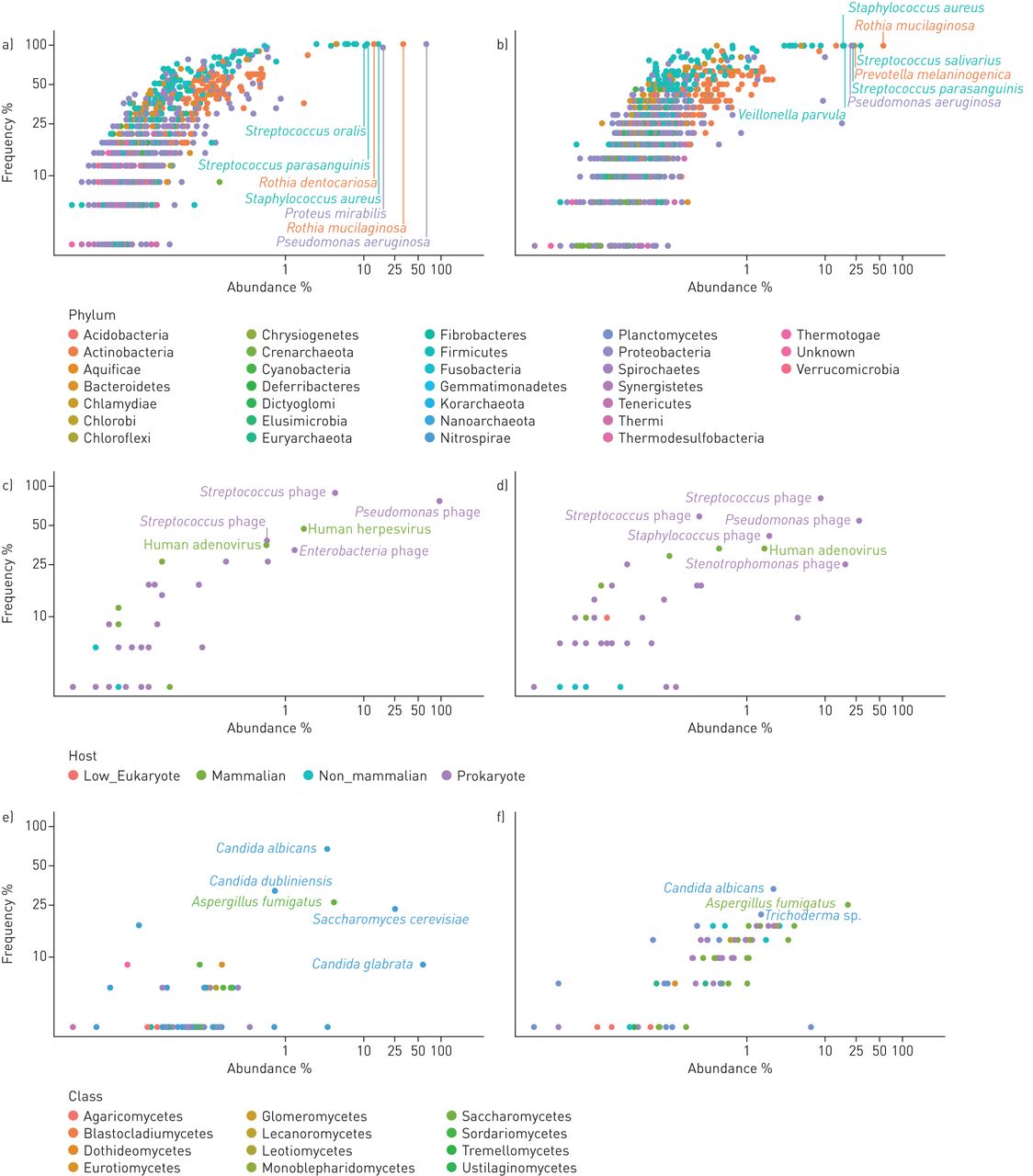
Frequency and abundance of microbial species in the cystic fibrosis (CF) lower airways metagenome. Each dot in the double logarithmic graphs depicts the identification frequency of a species and its mean relative abundance in sputa collected from a, c, e) exocrine pancreas insufficient and b, d, f) exocrine pancreas sufficient CF patients. Abundance was separately normalised for a, b) bacteria, c, d) DNA viruses and e, f) moulds and fungi. The name of a taxon is depicted for the most common and most abundant species. Bacterial taxa belonging to the same phylum are indexed by matching colour. Viruses are differentiated by their host. Moulds and fungi are differentiated by class. Please note that the data points depict the relative abundance of individual species found in the whole dataset of all exocrine pancreas insufficient and sufficient samples.
Common bacterial phyla in sputum from people with PS cystic fibrosis and PI cystic fibrosis#
The lead pathogen P. aeruginosa was identified in all specimens from PI patients although six of them had been classified as P. aeruginosa negative according to the clinical records. This finding suggests the ubiquitous presence of P. aeruginosa in respiratory secretions of PI CF patients. However, in all specimens taken from these misclassified patients P. aeruginosa was only present at low abundance on average in 0.02% of all reads. When we processed 24 respiratory specimens from healthy volunteers by the same protocol, no P. aeruginosa sequences were detectable in 14 samples and <10 reads could be assigned to P. aeruginosa in six samples (table S5). This data strongly indicates that the P. aeruginosa sequences detected in the CF specimens were retrieved from the patients' airways and did not originate from any contamination during processing of the sample. Our metagenome data point to an acquisition of P. aeruginosa early in life. The initially minute reservoir may then expand to clinically relevant numbers during exacerbations and/or at some stage of chronic airway remodelling.
The cladograms in figure 3 focus on the bacteria which made up the top 95% of the cumulative metagenome population of PI (figure 3a) and PS CF patients (figure 3b). The population in PI CF airways was dominated by pseudomonads and staphylococci followed by Veillonella, Streptococci, Prevotella, Rothia and other enterobacteriaceae as minor contributors. The spectrum of genera was similar in PS CF patients, but streptococci were more prominent and the population was more diverse and less skewed towards P. aeruginosa. Principal component analysis revealed a broadly scattering distribution of data sets retrieved from PS patients and strong clustering of data sets for the samples from PI subjects (figure 4) indicating that bacterial communities are more host-specific in PS CF and more disease-specific in PI CF.
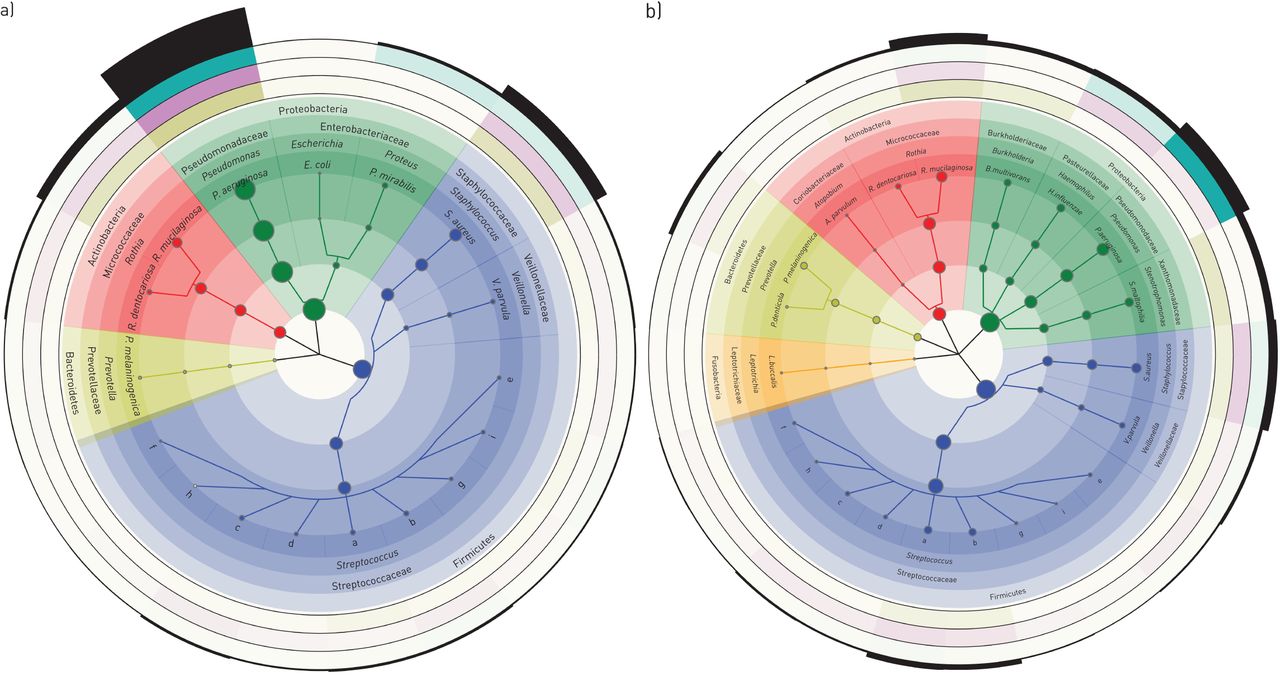
Taxonomic cladograms reporting the most abundant species contributing to the top 95% of the bacterial communities of the lower airways of a) exocrine pancreas insufficient and b) exocrine pancreas sufficient cystic fibrosis patients. Circle size is proportional to the log of average abundance of all analysed specimens. The height of the segments of the outermost ring indicates the relative average abundance of the respective genera and phyla in all age groups. The next three outermost circles indicate the relative average abundance of clades by colour intensity for adults (green), adolescents (purple) and children (olive). The analysis is based on 38 and 24 samples from exocrine pancreas insufficient and exocrine pancreas sufficient patients, respectively. Species of the genus Streptococcus are: a: S. parasanguinis; b: S. salivarius; c: S. oralis; d: S. mitis; e: S. pneumoniae; f: S. thermophilus; g: S. sanguinis; h: S. pseudopneumoniae; i: S. gordonii. The size of the dots indicates the mean of the average abundance of the three age groups. Within each age group, the mean of the relative abundance of all samples was calculated. Note the different evaluations of the dataset in figures 2 and 3.

Principal component analysis of the sputum metagenome data sets of exocrine pancreas insufficient (PI) and exocrine pancreas sufficient (PS) patients either a–c) not normalised or d–f) normalised to human DNA in the sample. The size of the concentration ellipses in probability was set to 0.95. Dim: dimension.
Individual microbial metagenome signatures and CF disease severity
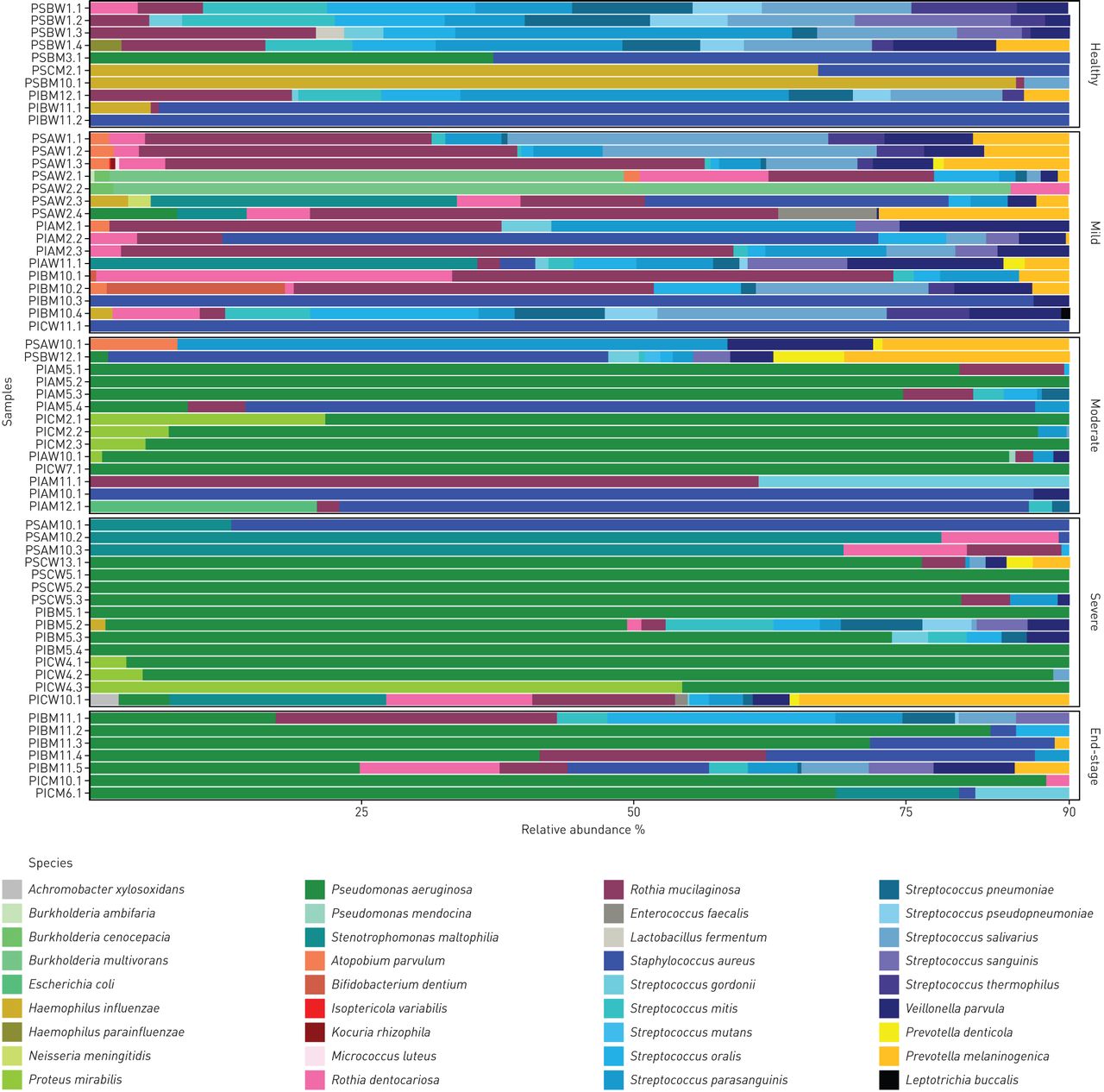
Whole metagenome analysis resolved the microbial signature of the individual patient. The spectrum ranged from normal flora in some healthy subjects with a normal lung clearance index via an intermediate stage when the normal community is perturbed by H. influenzae or S. aureus to a final stage of a low-diversity community dominated by P. aeruginosa (table S2, figure 5, figure S2) [28]. This shift from a normal highly diverse metagenome indistinguishable from that of a healthy subject to the CF typical, end-stage of an almost pure culture of P. aeruginosa was correlated in our patient cohort with disease severity, but not with age. As shown in figure 5, the diversity of the bacterial communities of the top 90% constituents decreased with increasing lung disease severity.

Composition of microbial communities in induced sputa of healthy and mild, moderate, severe or very severe (end-stage) lung disease affected individuals with cystic fibrosis. Data are presented in a stacked bar chart of relative abundance as fractions of total reads of the top 90% of species for each patient. The nomenclature of samples is explained in the supplementary material. Classification of disease severity is defined in the Methods section.
The metagenome of a CF patient was typically found to be made up of an individual signature of multiple, lowly abundant species superimposed by few disease-associated pathogens, such as P. aeruginosa and S. aureus, as major components (table S6). This phenotype became more obvious if we normalised the microbial reads to the human DNA in the sample. For example, as is illustrated in figure 6, a PS CF patient appeared to have an unrelated metagenome to that of a PI CF patient with chronic colonisation with P. aeruginosa if presented in bar charts as fractions of total microbial reads (figure 6a and b). However, after human DNA normalisation it can be seen that a quantitatively similar pattern of non-pathogenic species (“normal flora”) in the two subjects is overshadowed by P. aeruginosa in the PI CF patient, whereas no typical CF pathogens are detectable in the healthy PS CF patient (figure 6c and d). This normalisation clarifies some discrepancies in complexity between the microbiomes resolved by comprehensive culture-independent techniques and those based on culture-dependent diagnostics, the latter driven to detect disease-associated microbes and ignore the normal flora.

Bacterial sputum microbiomes of a, c) an exocrine pancreas insufficient (PI) cystic fibrosis (CF) patient who is homozygous for the most common cystic fibrosis transmembrane conductance regulator (CFTR) mutation p.Phe508del and b, d) an exocrine pancreas sufficient (PS) CF patient who is homozygous for the rare splice mutation in the CFTR gene c.1766+3 A-G. Data are presented as a, b) stacked bar charts of relative abundance as fractions of total bacterial reads or c, d) number of bacterial reads per human cell. Whereas the normalised bacterial microbiomes (a, b) suggest unrelated compositions of the bacterial communities in the respiratory tracts of the two patients, the normalisation per human cell (c, d) unravels similar colonisation modes with anaerobes in the two patients, which are overshadowed by the dominant Pseudomonas aeruginosa community in the PI patient. Tracings show original recordings of short circuit currents in rectal suction biopsies [22] to characterise the basic defect and CFTR function in the two individuals with CF. The horizontal arrow corresponds to a recording time of 30 min and the vertical arrow to a current of 10 µA·cm−2. Intestinal current measurements (ICM) were performed per protocol [22] by the addition of amiloride (1), indomethacin (2), carbachol (3), cAMP/forskolin (4), 4,4'-diisothiocyanostilbene-2,2'-disulfonic acid (DIDS) (5) and histamine (6). Carbachol, cAMP/forkolin and histamine induce chloride secretory responses seen in the ICM by signals in the upward direction. The tracings show either no (a, c) or intermediate (b, d) CFTR activity, indicated by the cumulative chloride currents (0 or 24 µA·cm−2) which implies that no functional CFTR is operating in the PI CF patient whereas substantial residual CFTR activity is present in the PS CF patient.
Clonal diversity of S. aureus and P. aeruginosa populations in CF lungs
The clonal composition of S. aureus and P. aeruginosa communities in CF sputa was determined from the frequency distribution of single-nucleotide polymorphisms in the metagenomes (figure 7, table S7). The P. aeruginosa communities consisted of one (16 sputa) or two major clone types (six sputa) making up 96–100% of the consortium. At least one further rare clone was detectable in 13 sputa. Within-clone variation was detectable for the most prevalent clone, which split up into two (12 specimens), three (six specimens), four (two specimens) or five (one specimen) discriminable clonal variants. The P. aeruginosa community remained rather stable in five of the six CF patients who provided serial specimens. A transient shift of the community structure was only observed for subject PIBM5 (figure 7).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clonal diversity of the Staphylococcus aureus and Pseudomonas aeruginosa communities in respiratory secretions. Metagenome samples with >40 000 species-specific reads were selected for analysis of single-nucleotide polymorphism (SNP) diversity in S. aureus (blue) and P. aeruginosa (green) sequences at reference genome positions covered by >10 reads. Data in the corner of each panel indicates the number of “mismatch” sequences, i.e. number of reads with at least one nucleotide substitution compared to the P. aeruginosa PAO1 and S. aureus Newman reference genomes. Reads sharing 100% sequence identity with the reference were designated as “matches”. The individual displays show the pooled number of “matches” (y axis) plotted in 1% intervals of the truncated ratio of pooled matches to the total number of reads in that interval (x axis). Clones are highlighted by a comparatively high number of sequences in neighbouring 1% intervals supported by many SNPs. Conversely, clonal variants are supported by only a few SNPs, thus the number of sequences in neighbouring intervals is comparatively low.
The S. aureus community consisted in all nine examined cases of one dominant (> 95% of all bacteria) and one minor clone. Two, three or four variants of the dominant clone could be discriminated in four, three and two samples, respectively.
Previous genotyping and subsequent genome sequencing of serial isolates had suggested that the CF lungs are chronically colonised with co-evolving clades of one or, less frequently, two or three unrelated clones [28–31], but our unbiased metagenome data indicate more diverse and more complex compositions of the S. aureus and P. aeruginosa populations in CF airways. Diversity of the S. aureus and P. aeruginosa communities is generated by intraclonal diversification [28, 30, 32, 33] and co-colonisation with unrelated clones.
To identify the genotype of the dominant S. aureus and P. aeruginosa strains within the frame of published typing schemes, the metagenome sequences were searched for matches with a multi-marker array for P. aeruginosa [34] and the multilocus sequence typing database for S. aureus [35]. Four out of the 10 analysed P. aeruginosa strains belonged to ubiquitous clones in the global P. aeruginosa population [34] and two pairs of 13 S. aureus strains were assigned to the common clone type ST7 and the pandemic methicillin-resistant S. aureus lineage ST22 [35], respectively.
Mutations in resistance genes to antimicrobial chemotherapy
The chronic airway infections in CF are treated by chronic or intermittent antimicrobial chemotherapy, at least on the occasion of a pulmonary exacerbation, often accompanied with the emergence of multidrug-resistant bacteria as the unwanted side-effect [1, 2]. We searched the S. aureus and P. aeruginosa sequences in the metagenomes for non-conservative coding variants in targets of anti-infectives and/or mediators of antimicrobial resistance that have a frequency of <20% in their pangenome (table 3). Mutations in the P. aeruginosa genomes affected genes that are known to be prone for mutation during antipseudomonal chemotherapy [36] but, in the case of S. aureus, the mutations also emerged in the gyrase-encoding gyr loci [37], which are the targets for fluoroquinolones. The patients' clinicians had never prescribed fluoroquinolones as anti-staphyloccocal chemotherapy. Besides the improbable cross-infection with a resistant strain, the treatment of concomitant infections by P. aeruginosa with a fluoroquinolone would be the most likely explanation for the collateral mutations in the S. aureus gyr genes. This example demonstrates the power of non-selective metagenome sequencing to detect genetic variations in traits of interest such as drug resistance, virulence or lifestyle.
Non-synonymous sequence variants of antibiotic resistance genes in Staphylococcus aureus and Pseudomonas aeruginosa populations of cystic fibrosis sputum metagenomes#
Discussion
Deep metagenome sequencing revealed a large repertoire of viruses, moulds, fungi, archaea and bacteria in the CF lung habitat. The lower airways metagenome of a CF patient was typically found to be made up of an individual signature of multiple, lowly abundant species superimposed by few classical CF pathogens, such as P. aeruginosa and S. aureus, as major components. This phenotype became more obvious if we normalised the microbial reads to the human DNA in the sample. This presentation of data in terms of absolute abundance of microbes, as shown in figure 6, was more similar to the outcome of routine culture-dependent diagnostics that just communicate disease-associated aerobes than the common output format of microbiomes (figure 5), which normalises all patients' samples to 100% irrespective of the absolute contents of microbes in the respiratory secretions [3–10]. In other words, the outcome of culture-dependent and culture-independent analysis of CF respiratory specimens is more similar than discordant modes of presentation may suggest.
The paediatric CF microbiome has been shown to be more diverse than that of CF adults indicating that there may be a time window for therapeutic intervention, which maintains diversity while reducing total bacterial load [7, 38]. Our study now shows cases of young CF adults who still have a healthy microbial metagenome (samples from subjects PSBW1 and PIBM12 (table S2) compared with those from healthy controls (table S5)). All study participants have been regularly seen since the age of diagnosis by a dedicated team of CF caregivers suggesting that continuous surveillance and intervention, if indicated, could prevent or decelerate progressive CF lung disease in some, but not all subjects.
Consistent with literature reports [39, 40], the lead CF pathogen P. aeruginosa became a major member of the microbial community in subjects with compromised lung function. Mucus plugging, airway remodelling, micro-colony and biofilm formation will then drive the regional isolation of the microbial metagenome [28, 41]. Considering this spatial heterogeneity, all sputa were collected by autogenic drainage in order to retrieve metagenomes that are representative for the whole lung.
Within-clone evolution of major clones is thought to trigger the adaptation of S. aureus and P. aeruginosa to the environment of the CF lungs [17, 28, 30, 31]. Our metagenome study now demonstrates that this concept does not cover the whole scenario. The S. aureus and P. aeruginosa populations do not only consist of one to three major clones, but also of rare clones. These infrequent clones constitute a low copy genetic resource which could rapidly expand as a response to habitat alterations, such as antimicrobial chemotherapy or invasion of novel microbes. Thanks to the high accuracy of sequencing-by-ligation in the colour space of 99.943% (see Methods section) we could resolve the clonal diversity of S. aureus and P. aeruginosa in CF airways. The error rates of the more often used sequencing-by-synthesis or single molecule real-time sequencing technologies are too high to reliably detect infrequent sequence variants, which may explain why minor constituents of polymicrobial communities at the rank of strains and clone types have not yet been reported in the literature.
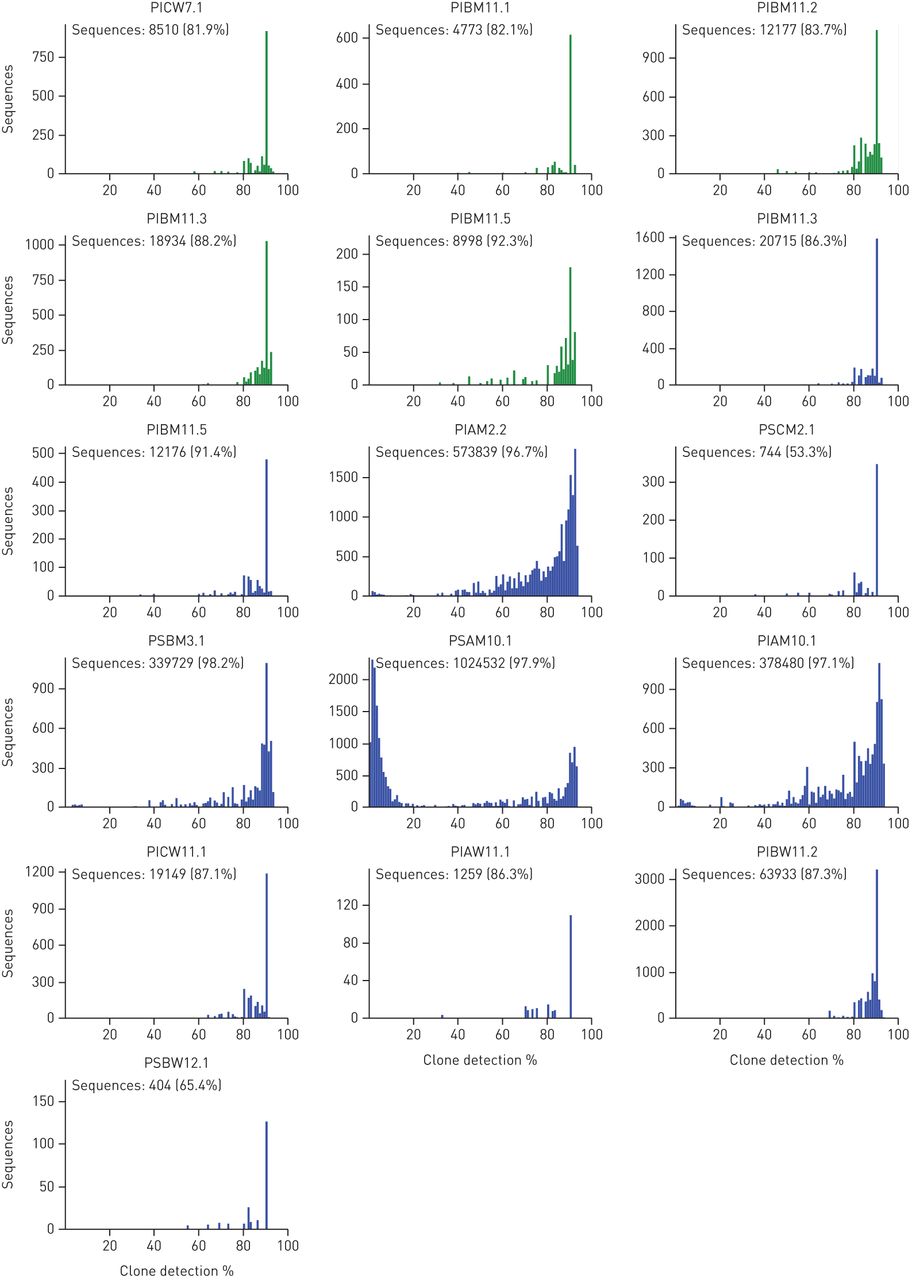
Metagenome sequencing generated quantitative and unbiased data about microbial diversity in CF lungs. Extensive culture-enriched profiling of the CF airway microbiome identified families of bacteria in CF sputa that were not detected by parallel 16S rDNA sequencing [42]. These biases of 16S amplicon sequencing do not apply to metagenome sequencing. Knowing that metagenome sequencing discerned, on average, one order of magnitude more organisms at the species level than 16S rDNA analyses [42], we consider any culture-enriched molecular analysis of CF sputum microbes to be dispensable if a metagenome approach is pursued. However, one should bear in mind that the sensitivity of the detection of rare members depends critically on the total number of microbial read sequences (figure S3). Unless one is interested in specific features, such as the spectrum of sequence variants in loci of interest, about half a million microbial reads are sufficient to provide a comprehensive metagenome analysis of taxa in CF airways.
Acknowledgements
P. Moran Losada and B. Tümmler conceived the study. M. Dorda, S. Hedtfeld, S. Mielke, A. Schulz and L. Wiehlmann performed the experiments. P. Moran Losada and P. Chouvarine wrote scripts. P. Moran Losada, P. Chouvarine L. Wiehlmann and B. Tümmler analysed the data. The manuscript was written by P. Moran Losada and B. Tümmler.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Support statement: This study was supported by funds from the Bundesministerium für Forschung und Technologie, German Center for Lung Research (DZL), and Mukoviszidose e.V. (project 1206). P. Moran Losada was supported by the Hannover Biomedical Research School (Hannover, Germany) and the Center for Infection Biology (Hannover). Funding information for this article has been deposited with FundRef
Conflict of interest: Disclosures can be found alongside this article at openres.ersjournals.com
- Received December 8, 2015.
- Accepted March 4, 2016.
- Copyright ©ERS 2016
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










