





Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease with poor prognosis, which is characterised by destruction of normal lung architecture and excessive deposition of lung extracellular matrix. The heterogeneity of disease progression in patients with IPF poses significant obstacles to patient care and prevents efficient development of novel therapeutic interventions. Blood biomarkers, reflecting pathobiological processes in the lung, could provide objective evidence of the underlying disease.
Longitudinally collected serum samples from the Bosentan Use in Interstitial Lung Disease (BUILD)-3 trial were used to measure four biomarkers (metalloproteinase-7 (MMP-7), Fas death receptor ligand, osteopontin and procollagen type I C-peptide), to assess their potential prognostic capabilities and to follow changes during disease progression in patients with IPF.
In baseline BUILD-3 samples, only MMP-7 showed clearly elevated protein levels compared with samples from healthy controls, and further investigations demonstrated that MMP-7 levels also increased over time. Baseline levels of MMP-7 were able to predict patients who had higher risk of worsening and, notably, baseline levels of MMP-7 could predict changes in FVC as early as month 4.
MMP-7 shows potential to be a reliable predictor of lung function decline and disease progression.
Abstract
MMP-7 is a predictive biomarker in IPF http://ow.ly/c69Y309aO4R
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with a high mortality rate and limited treatment possibilities. Two recently approved drugs, pirfenidone (Esbriet; Genentech, San Francisco, CA, USA) and nintedanib (Ofev; Boehringer Ingelheim, Ingelheim, Germany), were shown to reduce the rate of decline in forced vital capacity (FVC) over 52 weeks in patients with IPF who had mild to moderate impairment in pulmonary function tests (PFTs), but neither is a cure [1]. Recent data from pre-specified subgroup analysis of pooled data from phase 3 trials suggest that these compounds show benefit across IPF subpopulations, although it remains to be firmly demonstrated whether this can be generally extended to patients with more severe impairment in PFTs [2, 3].
Accurate diagnosis and evaluation of disease stage has an impact on the prognosis, optimal choice of treatment and monitoring of patients with various diseases. Unfortunately, there are currently no standardised definitions for IPF staging, and although PFT thresholds were commonly used in clinical trials to separate patients with mild to moderate disease from those with severe disease (FVC threshold: 50–55% predicted, and diffusing capacity of the lungs for carbon monoxide (DLCO) threshold of 35–40%), this classification has limited prognostic value for the clinical course alone [4]. More recently, a personal clinical and molecular mortality prediction index (PCMI), combining physiological, clinical and biomarker variables, has been proposed to estimate individual risk in patients with IPF [5]. The inclusion of a molecular biomarker in this scoring system was thought to have an improved mortality prediction value compared to the GAP (gender, age and physiology) scoring system that combines mainly physiological and clinical variables [6, 7]. Although the experience gained from those different scoring systems may help to support early diagnosis and prognosis of patients with IPF [8], the heterogeneity of the IPF patient population, which manifests in the presentation of different disease courses, remains a significant obstacle to offering optimal individual patient care, and it hampers efficient development of novel treatments. Disease-relevant biomarkers with reliable prognostic value could be included in a risk-scoring system to enable personalised treatment and to facilitate patient stratification for the development of novel treatments for IPF subpopulations [4].
IPF is characterised by a progressive remodelling of the lung, which is associated with loss of functional lung architecture and excessive deposition of extracellular matrix (ECM). Therefore, ECM proteins, among others, have been explored previously as indicators of disease activity, and as potential diagnostic and prognostic biomarkers in patients with IPF. However, the majority of previous biomarker studies suffered from small sample sizes and lack of longitudinal blood sampling to reach conclusive data. It is thus not surprising that routine clinical application of biomarkers in IPF has not yet reached maturity [9, 10].
During the randomised, prospective, placebo-controlled Bosentan Use in Interstitial Lung Disease (BUILD)-3 trial [11], serum samples and clinical data were collected at baseline, 4 months after treatment initiation and at end of study (EOS) from patients with IPF of less than 3 years' duration (i.e. with no or minimal honeycombing on baseline high-resolution lung computed tomography (HRCT)). The particular patient population characteristics, the high quality of the clinical data, the substantial sample number and the fact that longitudinal serum sampling was performed in combination with clinical measurements, makes the BUILD-3 sample collection a particularly valuable asset to explore biomarkers with diagnostic or prognostic potential and, furthermore, to look for biomarkers that would monitor disease progression. In the present study, we assessed the potential prognostic potential of four previously described biomarkers, matrix metalloproteinase-7 (MMP-7), Fas death receptor ligand (Fas ligand (FasL)), osteopontin (OPN) and procollagen type I C-peptide (PICP), which have been reported as associated with known pathobiological processes in IPF [12–15]. MMP-7, one of the best described biomarkers in IPF so far, is a member of the extensive MMP family, with the smallest molecular weight (247 amino acids). MMP-7 is expressed in alveolar macrophages and in alveolar and bronchiolar epithelial cells, and is known to degrade several ECM components. MMP-7 levels were shown to be elevated in serum and bronchoalveolar lavage (BAL) fluid from patients with IPF compared with healthy controls (HCs), but the marker is not sufficiently specific for the diagnosis of IPF [10, 16]. MMP-7 plasma levels were shown to be negatively correlated with FVC and DLCO decline [14], and helped mortality prediction together with FVC and DLCO (PCMI) [5]. This suggests that MMP-7 has potential to be a prognostic disease biomarker. In a larger study, including 438 patients, MMP-7 was shown to be an independent predictor of survival in a combined model that incorporated clinical parameters (sex, PFTs) and MUC5B genotype [17]. Osteopontin (OPN), which histochemically colocalizes with MMP-7 in the lungs of patients with IPF, is a mediator of inflammation and fibrosis [13]. Interestingly, MMP-7 expression and activity is induced by OPN, while OPN is cleaved and activated by MMP-7 [18]. OPN is elevated in the BAL fluid and plasma of patients with IPF compared with HCs, but OPN has limited diagnostic potential, as it is also elevated in other interstitial lung diseases (ILDs) [19]. OPN gene expression is more pronounced in the lung of patients with IPF with progressive versus those with stable disease [20]. FasL belongs to the tumour necrosis (TNF)-1 family, and induces pro-apoptotic signalling in the target cell upon binding to the Fas death receptor. Past studies have shown increased levels of soluble FasL in BAL fluids from patients with IPF, and FasL increased apoptosis of epithelial cells in vitro [12, 21–23]. Recently, a potential anti-fibrotic effect of FasL-positive immune cells has been proposed [24]. PICP is known to be an early marker of collagen synthesis in cardiac and hepatic fibrosis [25, 26].
Our results indicate that of the four biomarkers that we selected for analysis in serum samples, only MMP-7 represents a potentially reliable prognostic marker of IPF at time of diagnosis and one that can predict the deterioration of lung function during the follow-up period.
Materials and methods
Study subjects and serum samples
Serum samples and clinical data were collected from participants in BUILD-3 (NCT 00391443), a multicentre, prospective, randomised (2:1), double-blind, placebo-controlled, event-driven, morbidity-mortality trial of bosentan in adults with IPF of less than 3 years’ duration [11]. Approval was obtained from all relevant ethics committees and institutional review boards before study start, and patients provided written informed consent.
Serum samples were collected as part of the clinical protocol at baseline, 4 months and EOS. In total, 347 patients with IPF consented to serum sampling: 113 patients in the placebo arm and 234 patients in the bosentan arm. After collection, serum samples were aliquoted and frozen at −80°C until use. Samples that had signs of haemolysis were excluded for multiplex and singleplex assays, thus samples from 342 patients (113 patients in the placebo arm and 229 patients in the bosentan arm) were analysed in the multiplex assays. Samples from 213 patients (63 patients in the placebo arm and 150 patients in the bosentan arm) were available for additional analysis for the singleplex assays.
Healthy donor sera
Control sera were purchased from Bioserve (Beltsville, MD, USA).
The first group of age- and sex- matched controls (Controls-1, N=100), with no lung disease, was used to measure levels of MMP-7, FasL, OPN and PICP using the multiplex assay. Control-1 members were Caucasian, aged 50–76 (mean=62, sd=6.49); 70 were male and 30 female. The second group (Control-2, N=40), with no lung disease, was used in the singleplex assay to measure levels of MMP-7. Control-2 members were Caucasian, aged 60–68 (mean=64, sd=2.15); 21 were male and 19 female.
Clinical endpoints
The study design and clinical endpoints used for the BUILD-3 clinical trial have been described previously [11]. No significant difference between treatment groups was observed for the primary endpoint analysis of time to IPF worsening (a confirmed decrease from baseline FVC of ≥10% and DLCO ≥15%, or acute exacerbation or death up to EOS) [11]. Patient baseline demographic and clinical characteristics are provided in table 1.
Patient baseline demographic and clinical characteristics
Multiplex assay
Levels of MMP-7, FasL, OPN and PICP were assessed on a bead-based platform (BioPlex, Bio-Rad, Hercules, CA, USA), using a sandwich antibody-based assay (MMP-7, FasL and OPN) or an antibody competitive inhibition assay (PICP), as previously described [27]. As shown previously [28], the assay demonstrates <10% median inter-analyte interference, and the median intra-assay coefficient of variation, as assessed by 28 replicate serum controls, was 18%. The lower limit of detection was 7.93 pg·mL−1 for MMP-7, 2.63 pg·mL−1 for FasL, 25.53 pg·mL−1 for OPN, and 76.0 ng·mL−1 for PICP. All specimen identification and pipetting was performed by a barcode-enabled, high-speed pipetting robot (Tecan, Morrisville, NC, USA).
Singleplex assay for MMP-7
MMP-7 was quantified using a magnetic Luminex performance assay specific for human MMP-7 (#LMPM907; RnD Systems, Minneapolis, MN, USA). Serum samples were randomised, diluted five-fold in calibrator diluent and incubated in microtiter plates with colour-coded magnetic microparticles pre-coated with MMP-7-specific antibody. After removal of unbound material, an MMP-7-specific biotinylated antibody was used to bind the captured MMP-7, then a streptavidin–phycoerythrin conjugate was added, and after a final wash step the fluorescent signal was read on a Luminex 200 Reader (Thermo Fisher, Zug, Switzerland).
Statistical analysis
We performed Box–Cox transformations [29] to PFT and biomarker variables to normalise the residual distributions and equalise the residual variance across treatment groups and time points. Out-of-range analyte concentrations were treated as such in the computation of the likelihood function. Lambda parameters ranging from −2 to 2 in steps of 0.5 were allowed.
In the multiplex assay, for each analyte, out-of-range below (or above) concentrations were replaced with the minimum (or maximum, respectively) observed value minus (or plus, respectively) a hundredth of its interdecile range. In the singleplex assay, the detection limit for undiluted MMP-7 was 450 pg·mL−1; values below this were filtered. Missing PFT or analyte values were otherwise not imputed. Changes in biomarker levels between controls and patients with IPF were assessed on their Box–Cox transformed values using one-sided Student's t-test, assuming unequal variances. To assess the statistical significance of a change in analyte or PFT value over time, the Friedman test was applied to check if there was an overall difference between the groups. If the null hypothesis was rejected for the Friedman test, the Wilcoxon signed-rank test was performed to assess pairwise comparisons. For these tests, only patients with values at all three time points were kept (N=195). No correction for multiple testing was performed.
Kaplan–Meier curves were produced and log-rank tests performed to compare time with clinical worsening based on analyte concentration or PCMI index. The association of biomarker levels (concentration at baseline and fold change from baseline) or PCMI levels (low or high) with time to clinical worsening was tested using Cox proportional hazards models.
To test the capacity of baseline MMP-7 to predict FVC changes at 4 months and EOS, we used a linear regression analysis with the Box–Cox transformed baseline FVC value and MMP-7 concentration as independent variables, and the FVC value at the considered time point as dependent variable.
Statistical analyses were performed using R version 3.2.2 (R Project, Vienna, Austria).
Results
MMP-7 levels are increased in patients with IPF
Patients enrolled in the BUILD-3 trial consisted of a homogenous population of patients with IPF of less than 3 years' duration, with a baseline FVC of ≥50% and a DLCO of ≥30% of predicted values. Patients were diagnosed with usual interstitial pneumonia (UIP) using surgical lung biopsy, and were without extensive honeycombing at baseline, as measured by HRCT [11].
To investigate whether FasL, MMP-7, OPN and PICP were increased in patients with IPF and to follow the expression levels of these potential biomarkers over time, we determined their expression levels in the BUILD-3 serum samples that were collected longitudinally, and we compared all protein levels, irrespective of the timing of blood withdrawals, with matched HCs. To eliminate a theoretical confounding influence of bosentan treatment, only samples from the placebo arm were used in the initial analysis.
As shown in figure 1a, PICP levels in the placebo group were not significantly elevated versus those of HCs at any measured time point. The geometric mean serum PICP concentrations and p-values in comparison with the HC group (6.76 µg·mL−1) were as follows. IPF baseline: 6.67 µg·mL−1 (p=0.329); IPF 4 months: 5.81 µg·mL−1 (p=0.847); IPF EOS: 5.93 µg·mL−1 (p=0.760). FasL levels in the placebo group were significantly elevated versus HCs at EOS but not at baseline or at 4 months (figure 1b). The geometric mean serum FasL concentrations and p-values for the patients with IPF in comparison with the HC group (0.0340 ng·mL−1) were as follows. IPF baseline: 0.0218 ng·mL−1 (p=0.934); IPF 4 months: 0.0153 ng·mL−1 (p=0.993); IPF EOS: 0.0896 ng·mL−1 (p<0.0001). A similar pattern was seen with OPN serum levels in the placebo group, which were significantly elevated versus HCs only at EOS (figure 1c). The geometric mean serum OPN concentrations and p-values in comparison with the HC group (1.28 ng·mL−1) were as follows. IPF baseline: 0.764 ng·mL−1 (p=0.966); IPF 4 months: 1.56 ng·mL−1 (p=0.221); IPF EOS: 6.63 ng·mL−1 (p<0.0001). In contrast to the other three biomarkers that were measured in our cohort, MMP-7 levels were significantly elevated in patients from the BUILD-3 placebo arm at all time points compared with HCs (figure 1d). The geometric mean serum MMP-7 concentrations and p-values in comparison with the HC group (1.25 ng·mL−1) were: IPF baseline: 2.25 ng·mL−1 (p<0.0001), IPF 4 months: 1.97 ng·mL−1 (p<0.01) and IPF EOS: 2.64 ng·mL−1 (p< 0.0001).

Serum concentrations of procollagen type I C-peptide (PICP), Fas ligand, osteopontin (OPN) and matrix metalloproteinase (MMP)-7 in healthy controls (HCs) and in patients with idiopathic pulmonary fibrosis (IPF) assessed using a multiplex platform. Box plot analysis of serum concentrations of a) PICP, b) FasL, c) OPN and d) MMP-7, in HCs (n=100), and in patients with IPF at baseline (n=113), 4 months (n=102) and end of study (EOS) (n=72). One-sided t-test with unequal variance was used to compare analyte concentrations in HCs and patients with IPF for each analyte at each time point. ns: nonsignificant. **: p≤0.01; #: p≤0.0001.
We thus found that the levels of FasL, MMP-7 and OPN were all significantly elevated in patients with IPF in the placebo group at EOS, but except for MMP-7, no statistically significant increase between the IPF and HC groups was detected at the earlier time points. MMP-7 was thus the sole biomarker that showed a significant increase at all time points, and was therefore further analysed as potential prognostic biomarker.
MMP-7 levels increase over time and MMP-7 baseline levels predict lung function decrease
Based on the findings that MMP-7 levels in patients from the placebo group were increased at baseline versus HCs, we next included baseline samples from the bosentan-treatment group in our analysis and found that the combined samples showed significant (p<0.0001) elevation of serum MMP-7 (geometric mean IPF: 2.26 ng·mL−1, geometric standard deviation: 5.19, N=342) versus HC (geometric mean control 1: 1.25 ng·mL−1, geometric standard deviation: 2.62, N=100) (figure 2a).

Matrix metalloproteinase (MMP)-7 serum protein levels differentiate idiopathic pulmonary fibrosis (IPF) and healthy control (HC) groups 1 and 2 with multiplex and singleplex assays. Serum MMP-7 levels in patients with IPF was increased at baseline in both a) the multiplex assay (IPF, n=342) and in b) the singleplex assay (IPF, n=40) compared with HCs. Note that two different sets of HCs were used: Control-1 (n=100) and Control-2 (n=40). #: p≤0.0001.
Because multiplex assays may display lower sensitivity and quality in comparison with a technically validated singleplex assay [30] and because MMP-7 became the focus of our further investigations, we developed a singleplex MMP-7 assay to gain further confidence in our experimental findings. In a small feasibility study, using a randomly selected subset of patients with IPF and an independent control set, we confirmed the significantly (p<0.0001) elevated MMP-7 levels in patients with IPF at baseline (geometric mean IPF: 4.94 ng·mL−1; geometric standard deviation: 1.70; N=40) in comparison with a second, independent control group (geometric mean control 2: 2.25 ng·mL−1; geometric standard deviation: 1.69; N=40) (figure 2b). We next applied the singleplex MMP-7 assay to all serum samples (placebo and bosentan arms at all time points) for which sufficient aliquots remained for re-analysis (N=638). When comparing the results that were obtained for the patients involved in the feasibility study and the main study, we found a high correlation (r=0.9) (figure S1). This reproducibility gave us confidence that the assay was reliable and could be used for further investigations into the predictive role of MMP-7 in patients with IPF. Figure 3a shows serum MMP-7 levels of all patients who had blood sampling and clinical evaluation performed at three time points (195 patients: 60 placebo and 135 bosentan). Significant changes in MMP-7 levels were seen at 4 months and EOS compared with baseline (p<0.001 and p<0.0001 respectively).
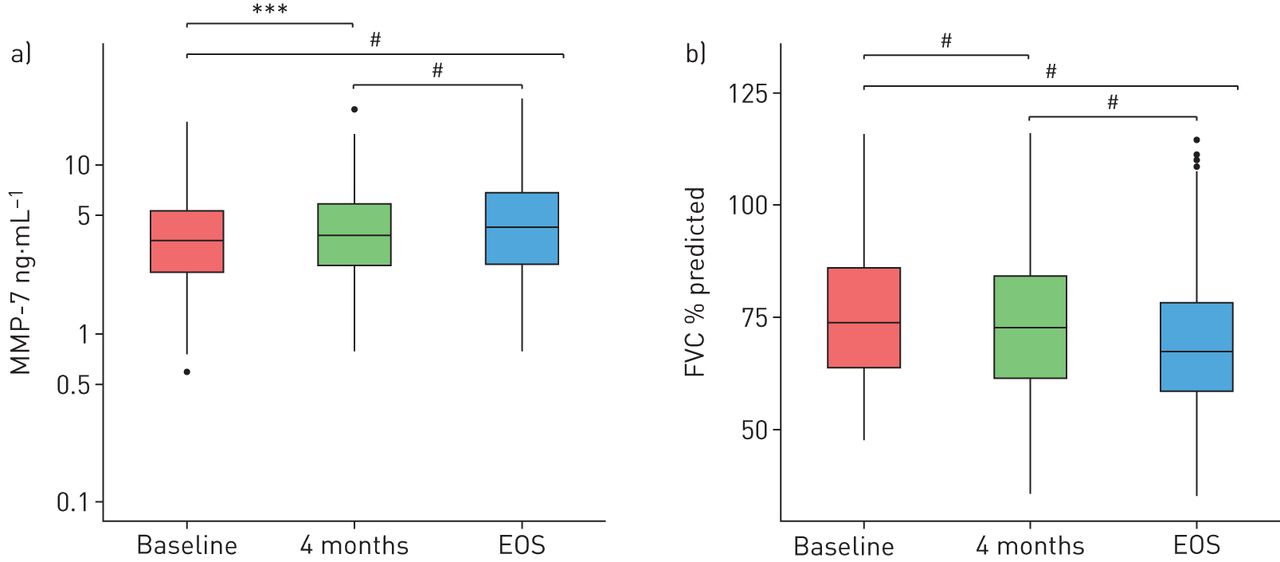
Increase in a) matrix metalloproteinase (MMP)-7 serum protein levels in patients with IPF was associated, in the same set of patients, with b) forced vital capacity (FVC) (% predicted value) decrease over time. EOS: end of study. ***: p≤0.001; #: p≤0.0001.
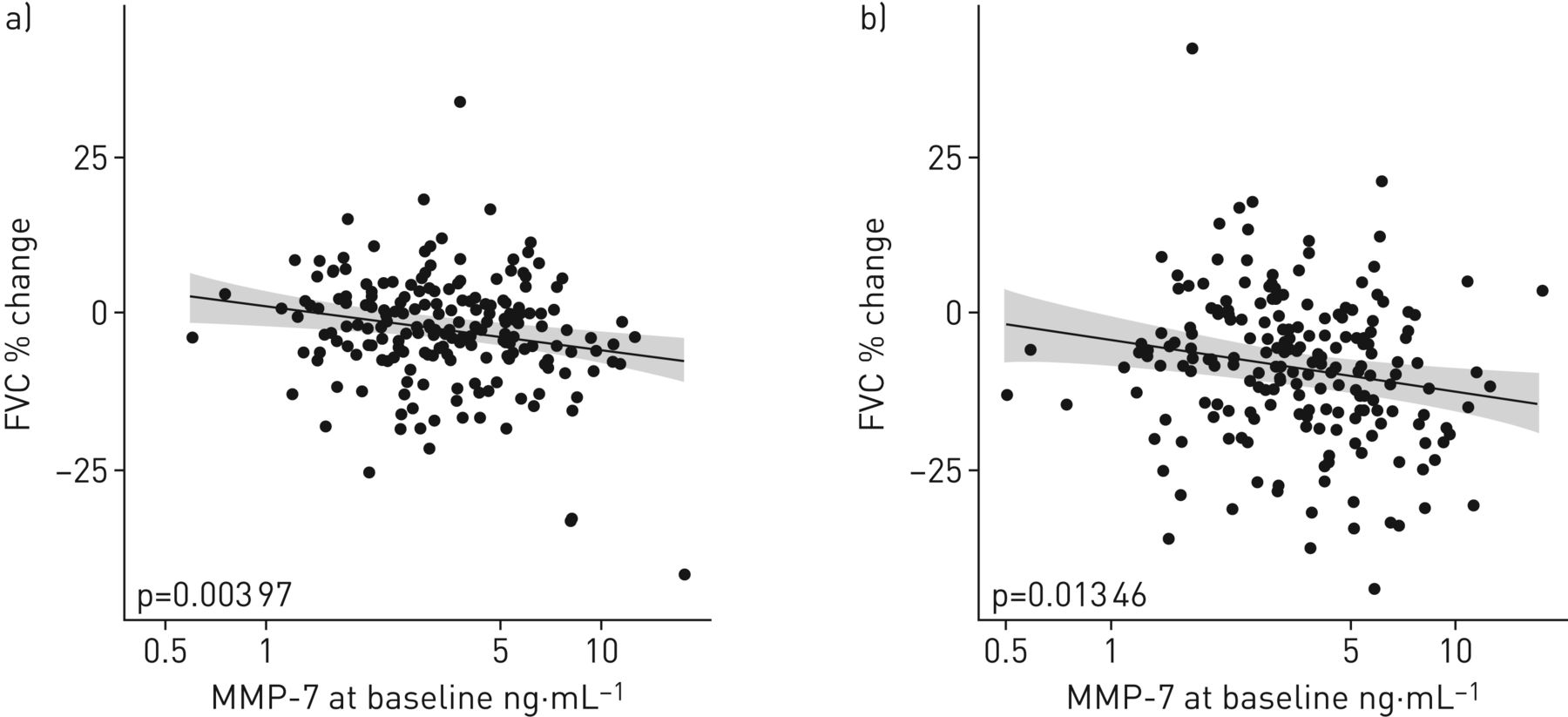
Significant changes in MMP-7 levels were also observed between 4 months and EOS (p<0.0001). The geometric mean serum MMP-7 concentrations at baseline, 4 months and EOS were as follows. IPF baseline: 3.48 ng·mL−1; IPF 4 months: 3.74 ng·mL−1; IPF EOS: 4.19 ng·mL−1. The corresponding mean FVC (%) levels at baseline (76.3%), 4 months (74.4%) and EOS (69.7%) are shown in figure 3b. It was notable that, concomitant with the significant longitudinal increases of MMP-7 serum levels, a statistically significant reduction in FVC (%) at 4 months and EOS versus baseline (p<0.0001 and p<0.0001, respectively) was seen. Furthermore, a significant reduction in FVC (%) was also seen from 4 months to EOS (p<0.0001) (figure 3b), suggesting that changes in serum MMP-7 levels followed disease progression. Additionally, baseline MMP-7 levels could predict changes in FVC between baseline and 4 months, as well as between baseline and EOS, as shown in figure 4 (p<0.01 and p<0.05, respectively). According to a Cox proportional hazard model (which included both the log2 MMP-7 levels at baseline and the log2 fold change between baseline and 4 months as covariates), patients with a two-fold change in MMP-7 levels between baseline and 4 months had a 1.9 times higher risk of worsening at any given time point (95% CI 1.2–3.0; p<0.005).

Scatter plot of the % change versus baseline in pulmonary function test results at a) 4 months and b) the end of the study versus the analyte concentration at baseline. The regression line (grey line) and 95% confidence interval (grey area) are represented. FVC: forced vital capacity; MMP: matrix metalloproteinase.
MMP-7 baseline levels predict patients at higher risk of subsequent disease progression
Next, the possibility that baseline MMP-7 levels could predict disease progression was explored. Using median MMP-7 values from all patients with singleplex data at all time points (N=208, table 1) as threshold, a significant difference in time to worsening was identified between patients with MMP-7 <3.8 ng·mL−1 (low) versus those with MMP-7 ≥3.8 ng·mL−1 (high) at baseline. Patients with high baseline MMP-7 levels had a 2.3 times higher risk of worsening at any given time point (hazard ratio (HR) 2.2, 95% CI 1.4–3.7; log-rank p=0.001; figure 5a).
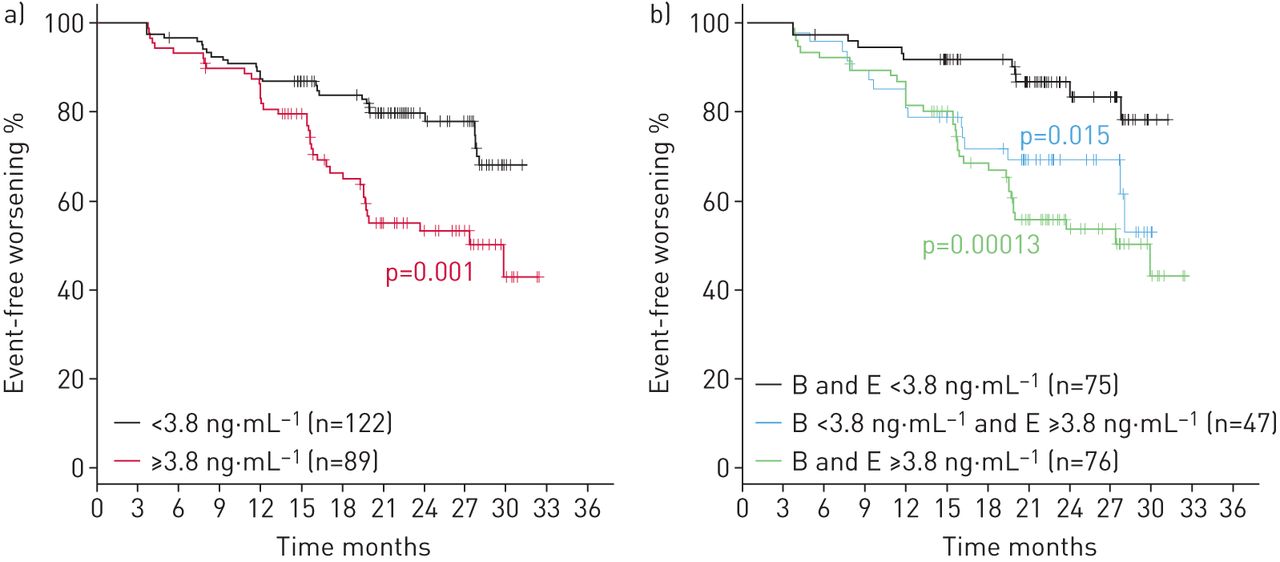
Kaplan–Meier estimate for clinical worsening in idiopathic pulmonary fibrosis patients a) by matrix metalloproteinase-7 median cut-off level at baseline (B) and b) by the median MMP-7 cut-off level when both baseline and end-of-study (E) values are considered.
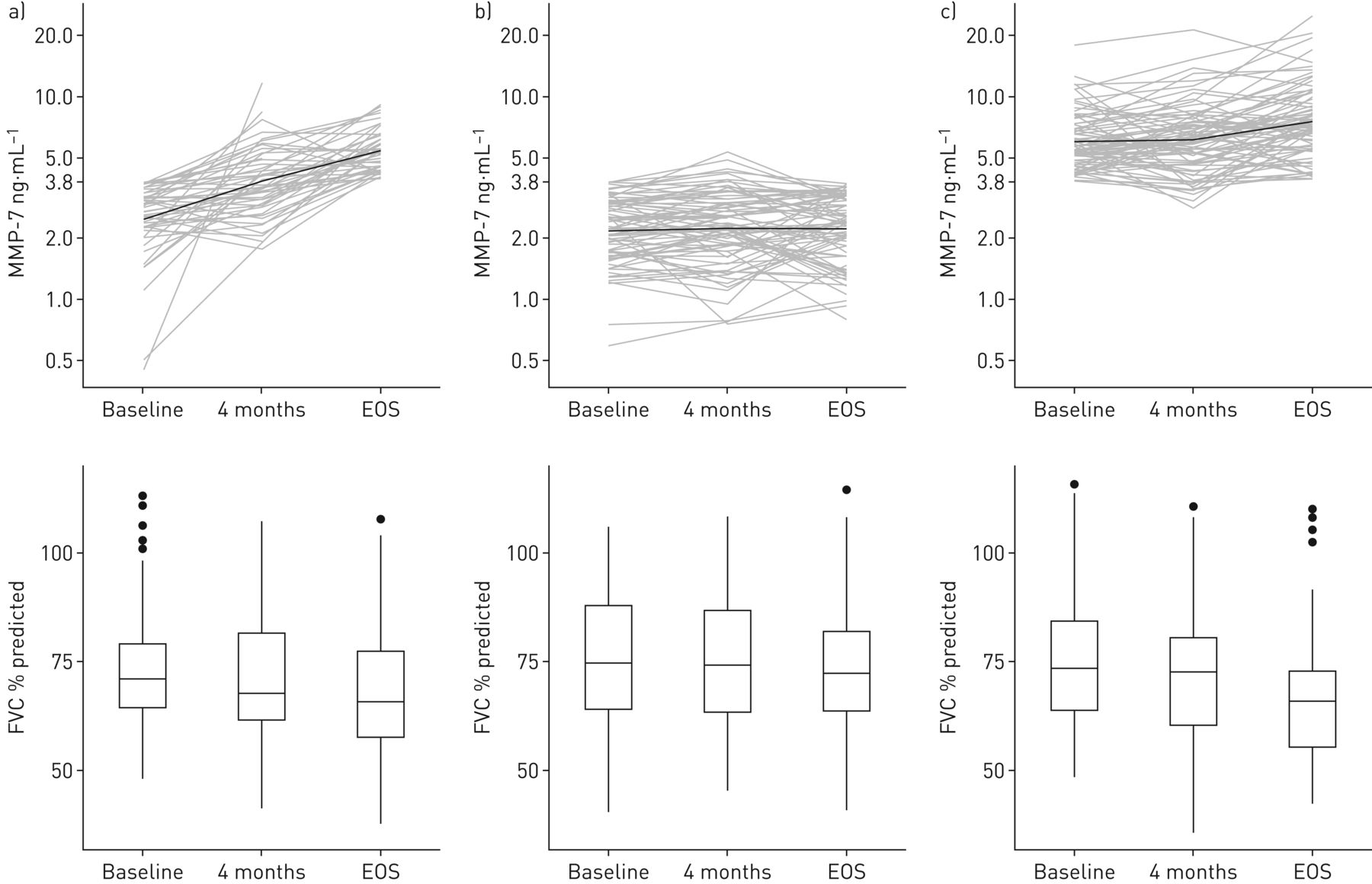
As we found that mean MMP-7 levels inversely correlated with the reduction of FVC over time (figure 3a and b), we performed a detailed analysis; we observed that patients who maintained low MMP-7 levels between baseline and EOS were more stable than patients with low MMP-7 levels at baseline and high MMP-7 levels at EOS, or patients with high MMP-7 levels at both baseline and EOS (HR 0.40, 95% CI 0.18–0.85, log-rank p<0.05; HR 0.29, 95% CI 0.15–0.57; log-rank p<0.001, respectively) (figure 5b). It is notable that patients with low MMP-7 levels at baseline and high MMP-7 levels at EOS usually showed increased MMP-7 levels at 4 months and a progressive decrease in FVC (%) over time (baseline: 76%, 95% CI 71–81%; 4 months: 73%, 95% CI 68–79%; EOS: 69%, 95% CI 63–74%; p<0.0001 between baseline and EOS) (figure 6a). Furthermore, patients with low MMP-7 at both baseline and EOS had no increase in MMP-7 at 4 months and had a lower FVC (%) decrease over time (baseline: 77%, 95% CI 73–80%, 4 months: 76%, 95% CI 72–80% and EOS: 74%, 95% CI 70–77%, p<0.001 between baseline and EOS) (figure 6b). By contrast, patients with high MMP-7 levels at both baseline and EOS showed no change in MMP-7 at 4 months and displayed a marked decrease in FVC (%) over time (baseline: 77%, 95% CI 73–80%, 4 months: 73%, 95% CI 70–77%; EOS: 66.%, 95% CI 63–70%; p<0.0001 between baseline and EOS).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in forced vital capacity (FVC) (% predicted) in relation to the evolution of matrix metalloproteinase (MMP)-7 level over time in the same set of patients. a) Baseline <3.8 ng·mL−1 and end-of-study (EOS) MMP-7 >3.8 ng·mL−1, b) baseline and EOS MMP-7 <3.8 ng·mL−1, and c) baseline and EOS MMP-7 ≥3.8 ng·mL−1.
Discussion
A striking feature in patients with IPF is the heterogeneity in disease progression, leading some patients to appear stable or to show a slow decline, whereas others decline rapidly with acute exacerbations and rapid deterioration of pulmonary functions [31]. This makes patient care management very challenging, and may have hampered successful development of novel disease-modifying drugs in the past. It is thus not surprising that much effort has been made to identify methods, including biomarkers, for reliable prognosis and to support the appropriate treatment regimens [32].
In this biomarker study, we employed serum samples from the BUILD-3 clinical trial to assess the prognostic capabilities of four previously reported putative IPF biomarkers: MMP-7, FasL, OPN and PICP. To our knowledge, BUILD-3 was the first large-scale phase 3 clinical trial in IPF to include longitudinal sampling for biomarker analysis. In our study, MMP-7 was the only biomarker that showed significantly increased levels at baseline, 4 months and EOS in BUILD-3 samples compared with HC samples. OPN and FasL were significantly elevated in patients with IPF versus HCs only at EOS, whereas PICP did not reach statistical difference at any time point. A recent paper [33] showed a statistically significant difference in plasma OPN levels between patients with IPF and HCs. It was therefore surprising in our study that OPN levels were not already clearly elevated versus HCs at the earlier time points. However, it is known that OPN can be proteolytically cleaved by thrombin during blood clotting and therefore OPN levels tend to be lower in serum than in plasma, which was confirmed in our study. We thus assume that differential OPN proteolysis, affecting assay sensitivity and variability, might account for the observed lack of statistical significance at baseline and 4 months, although a slight trend to higher OPN levels in patients with IPF was indeed visible at 4 months. For those reasons we did not investigate OPN further. However, it cannot be excluded that future studies, possibly using plasma samples, may be able to clarify if OPN also has potential as a disease progression biomarker.
By contrast, MMP-7 is a very robust biomarker in both plasma and serum, and was shown to be clearly elevated in patients with IPF compared with HCs in various studies. Furthermore and more importantly, when assessed longitudinally, there was a progressive increase of median MMP-7 levels and a concomitant decrease in median FVC (%). High baseline MMP-7 levels were predictive of a higher risk of worsening over time and could predict changes in FVC (%) as early as month 4.
An influence of treatment on MMP-7 levels or differential treatment response based on MMP-7 levels (data not shown) could not be demonstrated, which is in line with the reported lack of efficacy of bosentan in BUILD-3. This finding does not compromise the potential of MMP-7 to predict therapeutic responses in IPF. Staging systems for patients with a particular disease support the prognosis and may influence the choice of therapy [4]. Combining biomarker data and noninvasive clinical parameters has shown benefit in the care of patients with systemic sclerosis by allowing assessment of the risk for developing pulmonary arterial hypertension [34]. Thus, MMP-7 could be included in existing risk-scoring or staging systems. However, when we explored the possibility of risk scoring using baseline MMP-7 levels, we found that MMP-7 alone was comparable with the PCMI index that combines clinical and MMP-7 data (figure S2). Although the reported MMP-7 assay and the blood sampling source (plasma) that were used to generate the reported PCMI index [5] differed from the singleplex assay and the blood sampling (serum) used in our study, the cut-off values were comparable in range, and in both, a similar value above cut-off identified patients with higher risk of subsequent disease progression. Our data support the idea that measuring baseline MMP-7 levels using a validated and sensitive assay may provide highly relevant data to predict disease course.
Baseline assessment of MMP-7 has been previously shown to predict mortality in patients with IPF [5]; however, it was never longitudinally assessed to determine whether changes in MMP-7 levels follow disease worsening. Our study is the first to demonstrate that MMP-7 level changes over time predict subsequent disease progression and therefore identify patients at higher risk. MMP-7 measurements could therefore help to tailor the available treatment therapies to a patient or a group of patients with similar characteristics. Pirfenidone (Esbriet) and nintedanib (Ofev), two drugs that were recently approved for the treatment of IPF, were shown to reduce disease progression. In this context, MMP-7 levels at baseline may help to identify patients who would best benefit from those drugs, and longitudinal measurement of MMP-7 levels could be used as a supporting tool to monitor response to therapy [32].
This study has some limitations, as samples were taken only at baseline, 4 months and EOS, with no additional intermediate biomarker assessments and with missing data. All patients included in this study had IPF of less than 3 years' duration, with a baseline FVC of ≥50% and a DLCO of ≥30% of predicted values, and were without extensive honeycombing at baseline as measured by HRCT, thus conclusions cannot be extrapolated to patients with more advanced disease severity. The total number of deaths in the 211 patients investigated was seven; of these, five patients had baseline MMP-7 levels >3.8 ng·mL−1. The low number of deaths did not allow us to show a link between baseline MMP-7 levels and mortality (Fisher's exact test p=0.135). One additional limitation is that our study did not have a validation cohort, although previous work describing MMP-7 as a putative IPF biomarker supports our findings. However, it would be necessary to replicate our findings in an independent, prospective validation cohort, using a validated assay, to determine predictive MMP-7 threshold levels for wider application.
The major strength of our study is that we retrospectively measured biomarkers in a well-controlled clinical study that was designed to assess drug efficacy in IPF. The patient characteristics are representative of a well-controlled clinical trial and are therefore not hampered by the usual drawbacks encountered in general biobanking, including heterogeneity in patient characteristics, timing of sampling and nonstandardised lung function assessments. Our study is the result of a close collaboration between academic research and the pharmaceutical industry in a joint effort to improve patient care and to increase knowledge on this fatal disease. In the similar, recently communicated PROFILE study, it was shown that proteolytic ECM fragments, which are produced by enhanced MMP activity in the lung, could be detected in serum, were seen to increase over time and could predict survival [35]. In this context, it is reassuring to know that circulating MMP-7 is one of the resident lung MMPs involved in this process, and this further strengthens the significance of our findings. Further studies will be required to substantiate these recent biomarker findings and to identify the most suitable candidate for routine clinical use. The recent data from such large longitudinal studies, including our MMP-7/BUILD-3 data and the PROFILE study, bring the IPF field closer to a validated assay than ever before.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
FIGURE S1. Reproducibility of the MMP-7 singleplex assay. MMP-7 measurements done on the same IPF patient samples in feasibility study and main study were compared. 00074-2016_figure_S1
FIGURE S2. Kaplan-Meier estimate for clinical worsening in IPF patients split by the PCMI index cut-off (326) at baseline that optimized the p value of the log rank test. 00074-2016_figure_S2
Disclosures
Supplementary Material
Y. Bauer 00074-2016_Bauer
P. Cornelisse 00074-2016_Cornelisse
S. de Bernard 00074-2016_de_Bernard
I. Leconte 00074-2016_Leconte
A. Morganti 00074-2016_Morganti
O. Nayler 00074-2016_Nayler
S. Roux 00074-2016_Roux
E.S. White 00074-2016_White
Acknowledgements
The authors thank Geoffroy Bourquin, Annette Trebaul, Bérengère Renault, Solange Meyer, Hervé Farine, Julie Hoerner, Daniel Strasser, Oliver Peter and Peter Groenen for technical support, and Martine Clozel and Beat Steiner (all Actelion Pharmaceuticals Ltd, Allschwil, Switzerland) for helpful discussions and encouragement.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Support statement: The work of Y. Bauer, O. Nayler, S. de Bernard, P. Cornelisse, I. Leconte, A. Morganti and S. Roux was funded by Actelion Pharmaceuticals Ltd., and the work of E.S. White was funded by US National Institutes of Health grant R01 HL109118 and funding from the Drews Sarcoidosis Research Fund. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at openres.ersjournals.com
- Received July 6, 2016.
- Accepted December 31, 2016.
- Copyright ©ERS 2017
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










