





Abstract
Understanding the natural history of a disease is as important as knowing its cause(s) for effective disease prevention and treatment. Yet, our current understanding of the natural history of chronic obstructive pulmonary disease (COPD) is incomplete and often controversial. This article discusses the current gaps, and hence opportunities for research, in this field. In particular, it discusses the following six specific questions. 1) Is COPD a “single” disease? 2) Is COPD “only” a lung disease? 3) When does COPD begin or what is “early” COPD? 4) How does COPD “progress”? 5) How do we assess disease “severity”? 6) Can COPD be prevented (beyond smoking cessation) or its course be modified once detected?
Abstract
A new review series starts in ERJ Open Research: “Gaps in our understanding of COPD” http://ow.ly/CFSD30gpXs8
Introduction
This is the first of a series of articles that ERJ Open Research will publish on the gaps and opportunities currently existing in a variety of aspects of the so-called chronic obstructive pulmonary disease (COPD). They do not pretend to be in-depth reviews but rather the summary of a constructive interaction between two good, old friends with a long-time interest in COPD. The goal of the series is to identify gaps in knowledge, and hence opportunities for research, which may eventually stimulate the younger generation of respiratory clinician–scientists to delve deeper into them and, hopefully, get some answers that help to move the field forward and, more importantly, the many patients who suffer from COPD around the globe.
This first article in the series deals with a seemingly simple, but key, question: what is the natural history of COPD? The natural history of a disease is “the course it takes in individual people from its pathological onset (‘inception’) until its eventual resolution through complete recovery or death” [1]. Understanding the natural history of a disease is as important as knowing its cause(s) for effective disease prevention and treatment [1]. The metaphor of the “iceberg of disease” indicates that for every case seen in the clinic, there are likely to be many more with pre-clinical disease in the community, thus highlighting the importance of different prevention strategies (figure 1).

a) Biological onset (inception), pre-clinical and clinical phases of the natural history of a disease. Four different preventive strategies are also illustrated. Tx: treatment. b) Target population and goals of these four preventive strategies. Reproduced from https://www.med.uottawa.ca/sim/data/Prevention_e.htm
From these general considerations, several questions (thus gaps and opportunities) arise. 1) Is COPD a “single” disease? 2) Is COPD “only” a lung disease? 3) When does COPD begin or what is “early” COPD? 4) How does COPD progress? 5) How do we assess disease “severity”? 6) Can COPD be prevented (beyond smoking cessation) or its course be modified once detected? This article expresses our thoughts on these six topics.
Is COPD a “single” disease?
Needless to say, different diseases have different natural histories, so the first relevant question we should ask when discussing the natural history of “COPD” is whether it is, or is not, a “single disease”. According to the definition proposed by the Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 report, “COPD is a common, preventable and treatable disease that is characterised by persistent respiratory symptoms and airflow limitation that is due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases” [2]. However, respiratory symptoms (dyspnoea, cough and sputum production) and airflow limitation are not at all specific to COPD, and can occur in many other respiratory diseases. Furthermore, some individuals experience similar symptoms in the presence of normal spirometry [3, 4] and, likewise, there are individuals with emphysema as assessed by computed tomography (hence, clear evidence of lung pathology) who also have normal spirometry [5, 6]. On the other hand, COPD has been associated with a number of different causes, such as genetic abnormalities (e.g. α1-antitrypsin deficiency), cigarette smoke, exposure to biomass fuels, past infection of the lung, history of asthma and abnormal lung development, and in a minority, but still a sizable proportion of spirometrically detected cases of COPD, no causative agent may be identified [7, 8], albeit that it may be related to other respiratory conditions like asthma [9].
So, is COPD a “single” disease? We and others believe that the answer to this question is no [10–14]. Rather, we all think that COPD is a syndrome, defined as a set of medical signs and symptoms that commonly occur together and may be related to each other without necessarily tying them to a single identifiable pathogenesis [15]. For the purpose of this review, it follows that the different diseases that are included within the COPD syndrome are most likely to have different natural histories.
Is COPD “only” a lung disease?
So far, the natural history of COPD has been restricted to the description of changes over time of the degree of airflow limitation as expressed by the forced expiratory volume in 1 s (FEV1) [16], assuming COPD to be simply a disease that affects only the lungs. Yet, COPD is a complex and heterogeneous disease with different pulmonary and extrapulmonary components that do not necessarily progress in parallel [11, 17, 18], and are not always reflected by FEV1 [19]. For instance, cough and expectoration generally improve with smoking cessation [20] but their natural history among those individuals in whom it persists is unclear, even though mucus hypersecretion is known to increase the risk of rapid lung function decline [21], pneumonia [22], hospital admission [23] and death [24]. Likewise, exacerbations of COPD (ECOPD) occur more frequently with lower values of FEV1, but the relationship is weak [25] and the inter-ECOPD interval for severe exacerbations shortens with time [26]. Finally, the natural history of the extrapulmonary manifestations in COPD is largely unknown [17]. What we do know, however, is that many patients with COPD manifest important extrapulmonary manifestations that negatively impact on overall outcomes [2]. Amongst them are several comorbidities that occur more frequently in these patients than patients without a COPD diagnosis. Coronary artery disease, cardiac arrhythmias, systolic and diastolic heart failure, osteoporosis, depression and anxiety, gastro-oesophageal reflux, renal failure, interstitial pulmonary fibrosis, and lung cancer having have not only an increased prevalence, but their presence has been associated with increased risk of death [27]. Very important, because of its impact on the functional capacity of the patients, is the presence of low body mass index (BMI) in a significant proportion of patients and sarcopenia with peripheral muscle dysfunction, even in COPD patients with normal BMI [2]. An important gap in this area relates to the fact that whatever little we know, is derived from studies involving patients with cigarette-related COPD, where the toxins entering the blood stream reach all the organs of the body [28]. Furthermore, little is known about the nature of the interaction between the lung component of COPD and the extrapulmonary manifestations [29]. Are they the result of spillover from the process affecting the lung or are they simply different organ manifestations of common pathobiological processes? Do they occur in COPDs caused by agents different from cigarettes? Do they follow a synchronous path? Finally, emerging evidence suggests that some of the features considered to be consequences of COPD are actually contributory to the development and progression of the disease. An example is physical inactivity, which has been shown in some cohorts to predict faster decline in lung function [30, 31]. This is at an early stage of exploration but offers potential avenues for clinical trials designed to arrest disease progression.
When does COPD begin and what is “early” COPD?
The inception of a disease is not a firmly defined concept [1]. Biologically speaking, the natural history of a disease is sometimes said to start at the moment of exposure to a causal agent in an individual susceptible to it [32]. In fact, this has been the traditional approach to the natural history of COPD since Fletcher and Peto [33]. Assuming that smoking exposure was the “only” causal agent of COPD, Fletcher and Peto [33] proposed that some smokers (so-called susceptible smokers) suffer an accelerated rate of decline of lung function from a normal peak lung function value at the age of 25 years that eventually lead to COPD (figure 2). In other words, according to this model, COPD starts when a young individual starts to smoke, most often at adolescence [34]. This biological concept of disease inception supposes a pre-clinical period where the disease is “silent” and hopefully reversible. This is the principle that supports screening for many diseases, most importantly various cancers. Detection of this true “early” COPD is not yet possible but the development of biomarkers that include the detection of genetic variants for susceptibility for development of COPD is a priority [35]. The concept of disease inception can be extended to very “early” events that can occur during pregnancy, before birth [36–38].

Different lung function trajectories potentially relevant for the understanding of the natural history of chronic obstructive pulmonary disease. a) Lung function growth and development through the lifespan of a normal subject. b) Reduced maximally attained lung function. c) Reduced duration of the plateau phase of maximal lung function. Accelerated lung function could begin in early adulthood and be d) gradual, e) episodic (e.g. exacerbations) or f) could begin in late life. None of these trajectories are exclusive. Reproduced from [16] with permission from the publisher.
Lung development and growth is a complex and tightly controlled process [39]. Human lungs develop from the embryonic foregut as buds from the laryngotracheal sulci at 26 days of gestation [16]. These lung buds undergo dichotomous branching, forming approximately 18 generations of airways over weeks 16–26 of gestation [16]. Alveolar walls form by septation in a process that continues through childhood [16]. Following formation of alveolar units, the lungs grow in size and surface area as the thorax grows during childhood and adolescence. Maximal lung function is attained in young adulthood (earlier in females) and, in the majority of normal individuals, remains constant for a period of 10 years or so [34].
Pathogenic processes that can lead to COPD may occur during lung development and growth (figure 2). The studies by Barker et al. [37], which related birth weight to health outcomes in late life, established that being born small had a measureable effect on lung function and increased COPD risk, although the anatomical basis of this effect remains undetermined. Subsequent studies have also established a role for prematurity, maternal smoking and maternal nutrition as early life determinants of lung function that could contribute to COPD risk [38, 40]. These risk factors may explain the correlation between poverty and observed mortality rates from COPD and other lung diseases, including COPD in nonsmokers [41]. In childhood, both passive and active cigarette smoking can compromise lung growth leading to reduced maximally attained lung function [40]. Lung function growth is also compromised among children who developed bronchopulmonary dysplasia [38, 42]. Similarly, both reactive airways, while controversial, and childhood infections also appear to compromise lung growth [38]. A potential mechanism is suggested by studies in an early childhood model of asthma in the rhesus macaque. In this model, the development of asthma led to a reduction in the number of airway branches [16], a feature that is also present in humans with COPD [43]. Whatever the mechanisms, a variety of factors can result in reduced maximally attained lung function, which can increase the risk for COPD later in life (figure 2).
Clinically speaking, disease onset is customarily thought to begin when a previously healthy person develops symptoms [35]. Indeed, the concept that COPD only develops in 15–50% of persons exposed to cigarettes is based on this premise and constitutes the basis for the use of spirometry only in persons with symptoms or history of respiratory problems. In this context, there would be a phase of “early” COPD where the disease expresses itself with some symptoms (or not) and is associated with minimal airflow limitation [35]. Importantly, “early” COPD has to be clearly differentiated from “mild” COPD, which can also occur in the elderly [44]. “Early” is a concept related to time (age) whereas “mild” is a concept related to the severity of airflow limitation (FEV1) [45, 46].
How does COPD “progress”?
After attaining a maximum in young adulthood, in most individuals, lung function remains constant for about 10 years, after which it slowly declines at about 20 mL·year−1 in the healthy never-smoking individual [34]. Healthy females have a less steep rate of lung function decline through life than males [34].
In cigarette smokers, the duration of the “plateau phase” can be reduced and the decline begin earlier, whereas some nonsmokers can also experience a shortened plateau phase that could result in compromised lung function at an earlier age and increased risk of COPD [16] (figure 2). The traditional Fletcher and Peto model of COPD proposes that in “susceptible” smokers, this plateau phase is followed by progressive clinical worsening of symptoms, particularly dyspnoea and more limitation of airflow, until the disease becomes more severe and limiting (figure 2). This “progressive” model of COPD may not be so, since data from clinical trials and observational studies suggest that FEV1 decline trajectories may be set at a younger age and follow parallel paths [47], as we have discussed above. The ECLIPSE study showed that, over a 3-year follow-up period, lung function decreased excessively (>40 mL·year−1) in 38% of patients, normally (21–40 mL·year−1) in 31%, could be considered stable (−20–+20 mL·year−1) in 23% and improved (>20 mL·year−1) in 8% [48]. The same observation has been made in two other studies. In the Japanese Hokaido cohort, followed for 5 years, 50% of the patients had a normal rate of FEV1 decline while 25% of patients actually had increase in lung function [49]. Likewise, the BODE cohort that followed patients up to 10 years observed that close to 62% of patients had a normal rate of FEV1 decline and 9% of patients improved one or more stages in the spirometric Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification [50]. Therefore, the concept that COPD is always progressive and worsens universally is not tenable. If we are to have more impact on the “natural course” of COPD, we believe the disease has to be tackled upstream, at much earlier stages (i.e. younger ages) than the current paradigm of waiting for symptoms to screen for disease presence. This could allow identification of the individuals susceptible to a rapid progression of the disease in whom to implement novel therapies aimed at altering that progression. Although the Fletcher–Peto curve was proposed originally as a conceptual model [51], it has been often read as a representation of “the” natural history of COPD, despite that the same authors insisted that alternate natural histories were also possible (figure 2). Properly validated biomarkers of different lung function trajectories (other than spirometry) will be essential for the design of future COPD prevention and treatment trials [16].
How do we assess disease “severity”?
Traditionally, “COPD” severity has been equated to the severity of airflow limitation. However, although this is a key component of the syndrome, it is not the only one. Other elements that can contribute to COPD severity that need not be tightly related to the degree of airflow limitation include, among others, the level of symptoms, the presence of emphysema, the frequency of ECOPD and the co-occurrence of comorbidities [52], because all of these contribute to the health status and outcomes of the patient [53]. It follows that they need to be included in the assessment of the severity of COPD, besides FEV1, and that “disease progression” in COPD necessarily needs to be a multidomain construct that includes these different elements [53]. To that end, a number of multidimensional indices, such as the BODE [54] and ADO indexes [55], have been proposed. In a longitudinal study, the change in the BODE score better predicted poor outcome than changes in the FEV1 [54]. A close analysis of the different components of BODE (BMI, dyspnoea score, degree of airflow limitation and exercise capacity) showed that the FEV1 became less important a predictor of mortality as disease severity increased, and that changes in the functional capacity, as measured by the 6-min walk test, increased their predictive capacity [50].
Can COPD be prevented (beyond smoking cessation) or its course modified with treatment?
Since smoking has been traditionally considered the only causal agent of COPD, smoking avoidance [56] and/or quitting as early as possible [34] have been the only prevention strategies recommended. Yet, as discussed above, now we know that other conditions, including early-life events, infections, and exposure to other gases and particles, can also cause COPD. This consideration opens important windows of opportunity for preventing the disease.
There are four levels of disease prevention (figure 1), which should not be confused [57]. 1) Primordial prevention aims to avoid the emergence and establishment of the social, economic and cultural patterns of living that are known to contribute to an elevated risk of disease. It includes the environmental control of disease vectors (e.g. air pollution) and general predisposing factors (e.g. illiteracy). 2) Primary prevention aims to prevent the onset of disease (i.e. to reduce its incidence) and so involves interventions for risk reduction that are applied before there is any evidence of disease (e.g. vaccination and smoking avoidance). Primary prevention requires to know at least one modifiable risk factor and the capacity to modify it, and it can be directed to individuals or whole communities. 3) Secondary prevention aims to detect a disease in its earliest stages, before symptoms appear, and intervening to slow or stop its progression on the assumption that earlier intervention will be more effective, and that the disease can be slowed or reversed. Screening is central to secondary prevention because otherwise unrecognised disease can be identified by tests that can be applied rapidly and on a large scale to distinguish apparently healthy people from those who probably have the disease (e.g. spirometry). To be detectable by screening, a disease must have a long latent period during which the disease can be identified before symptoms appear, as may be the case in COPD. Finally, 4) tertiary prevention aims to arrest or reduce disease progression, once diagnosed (e.g. smoking cessation).
So, let us examine the question of whether COPD can be prevented beyond avoidance of smoking or its course modified with treatment from the perspective of these four different preventive strategies. 1) Primordial strategy: it is beyond discussion that promoting public health should be enforced for all human diseases, including COPD. 2) Primary prevention aims to prevent or reduce the occurrence (i.e. incidence) of COPD. Traditionally, this has been restricted exclusively to smoking avoidance, yet the identification of other important COPD risk factors may open new windows of opportunity for primary prevention. For instance, other potentially relevant exposures (e.g. biomass indoor cooking) must be avoided too. Similarly, increasing knowledge of the genetics of COPD (beyond α1-antitrypsin deficiency [58]) may facilitate the development of so-called P4 medicine (personalised, predictive, preventive and participatory) strategies [59]. Finally, if, as discussed above, early-life events can limit lung growth and development, and contribute to persistent airflow limitation later in life [9, 40, 60, 61], these should be considered as potential therapeutic targets or, currently at least, important research goals. 3) Secondary prevention (i.e. screening for COPD in the general population) is a controversial topic. A recent task force of the US Department of Health and Human Services systematically reviewed the evidence for and against screening for COPD, and concluded that “there was no direct evidence available to determine the benefits and harms of screening asymptomatic adults for COPD using questionnaires or office-based screening pulmonary function testing or to determine the benefits of treatment in screen-detected populations” [62]. On the other hand, the 2017 GOLD report advocates active case finding (i.e. performing spirometry in patients with symptoms and/or risk factors, where the diagnostic yield for COPD is relatively high) but not routine screening spirometry in asymptomatic individuals without COPD risk factors [2]. Case finding (i.e. after the development of symptoms but before presentation to healthcare services) offers an important opportunity for intervention since treatment may have an impact on outcome. In any case, these recommendations may need to be reconsidered in view of the new evidence indicating that there are several trajectories leading to COPD in adulthood and that not all of them are related to exposure to traditional risk factors [60]. For instance, fortunately, survival of (extremely) prematurely born babies has improved extraordinarily in the last two decades [42]. These “survivors” are now in their teens or early adulthood. It is therefore conceivable that adult pulmonologists will see more and more of them with abnormal lung function in the years to come [63]. Whether or not they will be symptomatic is arguable since lung function reserve at this age may be still large enough and, almost invariably, individuals adapt their lifestyle precisely to minimise symptoms. Thus, the issue of when these survivors with suboptimal lung function should be identified and, eventually, treated needs to be addressed. Recent research has convincingly shown in three different cohorts that they constitute a group of individuals at high risk of earlier comorbidity and premature death [64]. Conceivably then, one may want to detect them as early as possible (childhood or school age) if there were an effective therapeutic intervention available but, even if not, strict measures to avoid active and passive smoking and other potentially damaging exposures, correct diet and nutrition, regular physical activity, and periodical monitoring seem advisable [38]. Measuring spirometry at the time that the individual applies for a driving license (generally in early adulthood) may be another alternative, although perhaps too late. In any case, this again identifies knowledge gaps and new opportunities for research. 4) Tertiary prevention (i.e. reducing disease progression): quitting smoking is currently advised [2] and there is evidence that the younger the individual quits, the better [34]. Conversely, there are no data on the natural history of “untreated” COPD, as treatments with meaningful benefits have been available for a while and it would be unethical to withhold them. Despite this, COPD treatment is generally regarded as not affecting lung function decline meaningfully. Yet, available evidence contradicts this general belief. For instance, in TORCH, both salmeterol and fluticasone propionate each resulted in a statistically significant reduction in FEV1 decline over 3 years of 13 mL·year−1 [65]; their combination was even more effective, although the difference versus placebo (16 mL·year−1) did not achieve the 20-mL·year−1 reduction generally regarded as clinically relevant (albeit somewhat arbitrarily chosen). Likewise, UPLIFT assessed whether tiotropium could slow lung function loss and, while there was no effect in the total population, a 6-mL·year−1 reduction in FEV1 decline was observed among those with milder disease [66]; concurrent treatment of subjects in UPLIFT with other medications, including inhaled corticosteroids and long acting β-agonists makes it difficult to conclude that there is no effect of treatment. Finally, in the recently completed SUMMIT trial, fluticasone fumarate and its combination with vilanterol resulted in a significant improvement in the rate of FEV1 decline of 8 mL·year−1 [67]. Although this difference might seem small, it should be considered that rate of FEV1 decline in the placebo group was 43 mL·year−1 and that this 8 mL represents a 20% difference per year, a value that if started “early”, could significantly impact on the ultimate degree of lung function loss. Thus, we believe that a nihilistic approach is not justifiable in view of currently available evidence. What may be needed above all is to plan studies addressing the specific type of disease progression that wants to be prevented or reduced. For instance, so far, patients have been recruited and randomised using only their baseline degree of airflow limitation. It would be desirable to stratify them by the rate of lung function decline, degree and progression of functional capacity limitation (addressing the systemic consequences of COPD) and number and type of comorbidities. This could lead to the finding of specific biomarkers of these phenotypes and the implementation of personalised treatment.
Finally, it is of note that, so far, the narrative of this text has focused on a biological model of disease progression, but there is also a natural history of the functional, social and psychological impact of the disease, in which symptoms restrict activity and participation with a consequent progression of deconditioning (the “disability spiral”), and thereby impact on employment and social isolation. Importantly, there is therapy (rehabilitation) that can interrupt this vicious cycle [68].
Conclusions
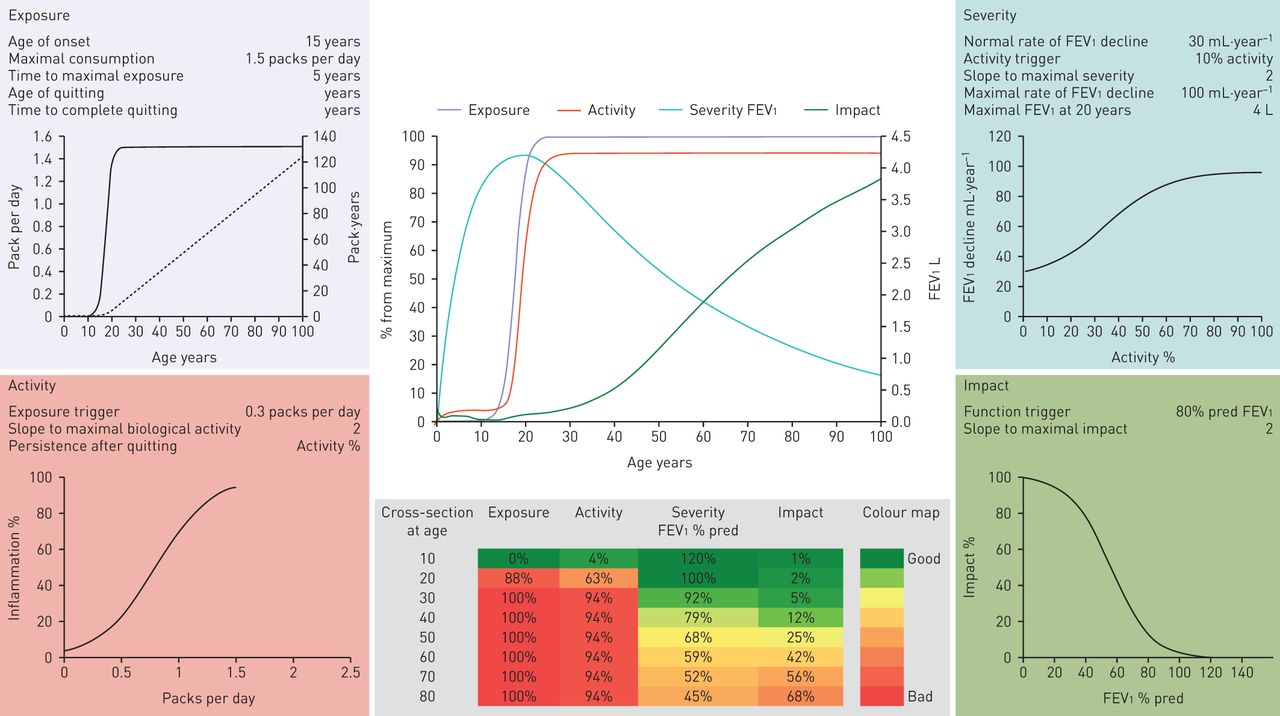
Herein, we have critically discussed the knowledge gaps and research opportunities related to the natural history of COPD. The following messages can be highlighted. 1) COPD is not a single disease; rather, it is a syndrome. Thus, there is no single COPD natural history. 2) What we call “COPD” today is the end result of a complex set of gene–environment interactions. They start during pregnancy, and continue after birth during infancy, adolescence, adulthood and old age. 3) Understanding the dynamic nature of COPD offers new windows of opportunity for prevention and treatment, provided that appropriate research is done. All these findings have to be incorporated into any new model(s) describing the natural history (or histories) of COPD, since each of these features is likely to have a natural history that may be quite independent of that of airflow limitation (table 1). In this context, we have recently proposed an integrative computational approximation to the natural history of COPD (the EASI model) that seeks to facilitate the investigation of the variability and determinants of different “individual natural histories” of COPD [69]. The EASI model integrates dynamically and represents graphically (figure 3) the relationships between exposure (E) to inhaled particles and gases (e.g. smoking), the biological activity (e.g. inflammatory response) of the disease (A), the severity (S) of airflow limitation (FEV1) and the impact (I) of the disease (breathlessness) in different clinical scenarios [69]. It is only a first step towards a better understanding of the natural history of COPD, without any current clinical application, but it can open new perspectives in the field. A fully workable spreadsheet is available in the online supplement of the original publication [69]. We invite you to experiment with, and hopefully enjoy, it.

{kind=link}
{kind=link}
{kind=link}
The EASI model structured around four modules (exposure (top left), activity (bottom left), severity (top right) and impact (bottom right)), each of which presents the parameter values used to calculate it and the relevant steady-state activation functions linking inputs from previous module (x-axis) to module outputs (y-axis). In the exposure module, the solid line indicates daily smoking exposure (packs per day; left y-axis) as a function of age (x-axis), whereas the dashed line corresponds to the cumulative smoking exposure (pack-years, right y-axis) of that theoretical individual. The EASI model also includes two central panels. The top one presents a longitudinal summary of the age-related trajectories of exposure, activity, forced expiratory volume in 1s (FEV1) (right y-axis) and symptoms. The bottom centre panel presents a heat map of these same four variables by decade of age. This particular example illustrates the EASI relationships for a susceptible continuous smoker. Reproduced with from [69] with permission from the publisher.
Selected markers that could be used to determine the natural history of patients with chronic obstructive pulmonary diseases
Disclosures
Footnotes
Number 1 in the series “Gaps in our understanding of COPD” Edited by A. Agustí and B. Celli
Conflict of interest: Disclosures can be found alongside this article at openres.ersjournals.com. Both authors are members of the Board of Directors of the Global Initiative for Obstructive Lung Disease (GOLD), but the opinions and proposals presented in this manuscript are their own and not necessarily those of GOLD.
- Received September 21, 2017.
- Accepted October 13, 2017.
- Copyright ©ERS 2017
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Is COPD a “single” disease?
- Is COPD “only” a lung disease?
- When does COPD begin and what is “early” COPD?
- How does COPD “progress”?
- How do we assess disease “severity”?
- Can COPD be prevented (beyond smoking cessation) or its course modified with treatment?
- Conclusions
- Disclosures
- Footnotes
- References
- Figures & Data
- Info & Metrics