





Abstract
Compartmentalisation of the respiratory tract microbiota in patients with different chronic obstructive pulmonary disease (COPD) severity degrees needs to be systematically investigated. In addition, it is unknown if the inflammatory and emphysematous milieux in patients with COPD are associated with changes in the respiratory tract microbiota and host macrophage gene expression.
We performed a cross-sectional study to compare non-COPD controls (n=10) to COPD patients (n=32) with different disease severity degrees. Samples (n=187) were obtained from different sites of the upper and lower respiratory tract. Microbiota analyses were performed by 16S ribosomal RNA gene sequencing and host gene expression analyses by quantitative real-time PCR of distinct markers of bronchoalveolar lavage cells.
Overall, the microbial communities of severe COPD (Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 3/4) patients clustered significantly differently to controls and less severe COPD (GOLD 1/2) patients (permutational multivariate ANOVA (MANOVA), p=0.001). However, we could not detect significant associations between the different sampling sites in the lower airways. In addition, the chosen set of host gene expression markers significantly separated COPD GOLD 3/4 patients, and we found correlations between the composition of the microbiota and the host data.
In conclusion, this study demonstrates associations between host gene expression and microbiota profiles that may influence the course of COPD.
Abstract
Associations of the host immune response and a disordered microbiota in patients with different COPD severity degrees http://ow.ly/h2mW30k9Nua
Introduction
It has been shown that patients with chronic obstructive pulmonary disease (COPD) not only suffer from infections that lead to exacerbations but, in general, show a disordered lung microbiota composition compared to healthy controls [1–4]. Nevertheless, data from stable COPD patients are still scarce and it is not yet fully understood whether there is a heterogeneity of the microbiota along the respiratory tract of patients with COPD, although microanatomic differences in bacterial communities within the same lung of subjects with advanced COPD have been suggested [2, 5]. Closely linked to the disordering of the microbiota is the change in the host immune status of patients with COPD. Specifically, innate airway immune cells, primarily macrophages, which are markedly increased in the airways of COPD patients, play a key role in the inflammatory processes in COPD [6]. Macrophages are a heterogeneous cell population endowed with plasticity in different disease states and extremes in polarisation, ranging from pro- to anti-inflammatory phenotypes [7]. In mice, prototypic M1- and M2-like activated macrophages may be distinguished, where M1-like macrophages are characterised as proinflammatory and M2-like macrophages as anti-inflammatory with release of interleukin (IL)-10 and phagocytic activity [8]. It has been suggested that proinflammatory macrophages predominate in COPD patients [6, 9], but the contribution of distinct phenotypes and the environmental factors that determine these phenotypes are still largely unknown.
In this study, we performed a cross-sectional analysis of patients (n=32) with different COPD lung functional severity degrees (grouped into Global Initiative for Chronic Obstructive Lung Disease (GOLD) grades 1–4, www.goldcopd.org), which were compared to controls (n=10). Different respiratory tract samples were obtained including both protected specimen brushes (PSBs) and bronchoalveolar lavage (BAL). The microbiota within the respiratory tract and the macrophage gene expression of the study participants were investigated. Our aims were to analyse differences in 1) microbiota profiles between the COPD severity degrees and non-COPD controls, 2) microbiota profiles at distinct sites of the respiratory tract, 3) macrophage gene expression in COPD patients and controls, and to understand 4) the relationship between altered microbiota and host macrophage phenotype.
Methods
Study design and sample collection
The study was approved by the institutional ethics board of the Canton of Bern, Switzerland. Between September 2013 and February 2016, participants with stable COPD (n=32) with bronchoscopies indicated for endoscopic lung volume reduction or suspected lung cancer were enrolled at the University Hospital of Bern. Details of recruitment of patients and controls as well as exclusion criteria can be found in the supplementary material. The following respiratory samples were obtained: the pharynx was sampled with PSBs; the trachea, main bronchus, lobar bronchi (upper, middle and lower on the right side; upper, lingula and lower lobe on the left side) were sampled during bronchoscopy with PSBs. Brushes were placed in tubes containing 1.4 mL sterile PBS. BAL was additionally obtained from either the middle lobe or the lingula as previously published [10]. Particular, state-of-the-art care was taken to minimise contamination during bronchoscopies as outlined in the supplementary material.
Sample procedures and 16S ribosomal RNA sequencing of the microbiota
DNA extractions were performed for all samples as described in detail previously and outlined in the supplementary material [11]. The protocols included a very stringent procedure to handle low-density material that involved the processing of negative controls and the exclusion of samples below a distinct DNA concentration threshold. This means that samples with <1.0 ng·μL−1 PCR product, corresponding to <1 pg·μL−1 bacterial DNA, were excluded (see supplementary material). Exclusion of samples from GOLD 3/4 patients was higher as compared to the other groups as bronchoscopy/BAL was more difficult to perform within these patients (e.g. to receive large quantities of BAL) and, therefore, fewer high-quality samples were received. BAL samples were additionally utilised for standard bacterial cultures. We also treated a subset of BAL samples (n=10) with propidium monoazide (PMA) (Biotium, Fremont, CA, USA) to remove dead bacterial cells before DNA extraction (see supplementary material for more details) in order to evaluate the consequences of prior PMA treatment for the microbiota results of our samples. PCR of the V3–V5 region of the 16S ribosomal RNA (rRNA) gene was performed using primer pair 341F/907R.
Sequence reads were analysed using the PyroTagger pipeline, which comprised quality filtering, removal of chimaeras, the definition of 97% operational taxonomic units (OTUs) and taxonomic assignment as described and outlined in the supplementary material [12]. Species richness, Shannon Diversity Index (SDI), and β-diversity were calculated in R version 3.3.0 (www.R-project.org) using the vegan package as described before and in more detail in the supplementary material [13, 14]. For the within-subject dissimilarity, the BAL from a subject was compared to all other respiratory samples within that subject. For the between-subject dissimilarity, the BAL samples from all subjects were compared pairwise. The project accession number for the sequence reads of this study is PRJEB20508.
Characterisation of BAL cell gene expression
Macrophages represent the predominant cells recovered by BAL and thus, characterisation of BAL cells gene expression is mainly a characterisation of macrophage gene expression [15, 16]. Quantitative real-time PCR was performed for tumour necrosis factor (TNF)-α, CXCL11, cyclo-oxygenase (COX)2, tissue inhibitor of metalloproteinase 1, platelet-derived growth factor (PDGF)-D, matrix metallopeptidase 12, dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin (DCSIGN), mannose receptor C (MRC)1, IL-1 receptor antagonist (IL1RN), IL-10 and indoleamine 2,3-dioxygenase (IDO) with the following innate immune functions in the BAL samples: inflammation, immunoregulation, scavenging and remodelling. The characterisation of the BAL cell gene expression was previously described and the procedure is detailed in the supplementary material [17].
Statistical and correlation analyses
For the PMA treatment analyses, where only two groups were compared, a paired t-test (for normally distributed data) or a Mann–Whitney test (for not normally distributed data) were performed. For the other analyses, where more than two groups were compared, two-way ANOVA tests were performed. Tukey's multiple comparisons tests were used to correct for the number of sampled sites (n=8) and COPD status (n=3). Relative abundances of bacterial families were log(Y+1)-transformed as they were not normally distributed. We analysed both the differences between the different sampling sites and the differences between patient groups. To test differential host gene expression, the data were log-transformed and Mann–Whitney tests were applied. In addition, a principal component analysis (PCA) was performed using prcomp from the stats package (scaled) routine in R. Correlation of macrophage gene expression markers with SDI and richness was done by Pearson correlation analysis. In addition, we performed univariate negative binomial regression modelling for the association of host gene expression with the relative abundances of bacterial families, as the latter were not normally distributed (details and results in the supplementary material).
Results
Study design, sample collection and quality control
We included a total of 42 participants (10 controls and 32 COPD patients) (table 1). COPD patients were stratified according to the degree of airflow limitation into patients with mild (GOLD 1) (n=6), moderate (GOLD 2) (n=8), severe (GOLD 3) (n=8) and very severe COPD (GOLD 4) (n=10). Patient characteristics are shown in detail in table 1. Patients with more severe airflow limitation (GOLD 3/4) had increased dyspnoea (modified Medical Research Council dyspnoea score ≥2), were more symptomatic (COPD Assessment Test score ≥10) and exacerbated more than once annually (table 1). In total, 187 samples from 32 COPD patients and 10 controls were analysed (table 1). After 16S rRNA sequencing, 172 (92%) samples passed all the quality checks (92%) (supplementary material). To achieve more statistical power, we subsequently grouped COPD GOLD 1 and 2, and GOLD 3 and 4. PMA pre-treatment for the removal of dead cells in a subset of BAL samples (n=10) did not have a significant effect on the microbiota composition (figure S1). Results of standard culture of BAL samples are shown in the supplementary material.
Description of the study participants and the number of samples per group and per sampling site
α-diversity values are decreased in GOLD 3/4 patients
We first investigated the α-diversity by calculating the sample richness and the SDI. There was no difference in species richness between controls and COPD patients, as well as no differences between the different sampling sites (figure S2A). The SDI, in contrast, was significantly decreased in COPD GOLD 3/4 patients as compared to controls and GOLD 1/2 patients (figure S2B) (two-way ANOVA; compared to controls: pharynx p=0.01, trachea p=0.02, middle lobe p=0.02 and BAL p<0.0001, respectively; compared to GOLD 1/2 patients: pharynx p=0.01 and BAL p=0.004, respectively).
COPD patients with early disease (GOLD 1/2) did not show changes in the SDI from normal subjects and, therefore, changes in SDI were restricted to advanced disease (GOLD 3/4).
Bacterial community comparisons and β-diversity measurements
Overall, COPD GOLD 3/4 patients clustered significantly differently compared to controls and GOLD 1/2 patients illustrated by hierarchical clustering based on bacterial families (figure 1) (permutational multivariate ANOVA (MANOVA), p=0.001). We observed increased Prevotellaceae and Acidaminococcaceae within healthy controls, which has been previously shown [18]. However, the bacterial communities are not significantly separated according to different sites along the respiratory tract (figure 1). We next investigated more thoroughly the differences in the microbiota along the respiratory tract (i.e. different sites), performing β-diversity analyses that included analysis based on 97% OTUs rather than bacterial families (figure 2a–c). With this analysis, we found a significantly different clustering only for the pharynx of COPD GOLD 1/2 and GOLD 3/4 patients (figure 2b and c) (permutational MANOVA; p=0.05 and p=0.04, respectively). We then calculated the dissimilarity between all samples of the respiratory tract within a subject (within-subject dissimilarity) and compared these values to the dissimilarity between BAL samples of different subjects (between-subject dissimilarity). There was a significantly lower within-subject dissimilarity compared to the between-subject dissimilarity in all groups (figure 2d–f) (Mann–Whitney tests, p<0.001 for all groups), indicating a more similar microbiota along the respiratory tract of a subject compared to the microbiota of BAL samples between different subjects. In addition, analysing the between-subject dissimilarity of the three groups (figure 2d–f) we found a gradual increase in dissimilarity by COPD severity degree (Mann–Whitney tests; p<0.0001, p<0.0001 and p=0.004). Therefore, these findings illustrate a more pronounced microbiota heterogeneity within COPD GOLD 3/4 patients compared to the controls and to GOLD 1/2 patients.
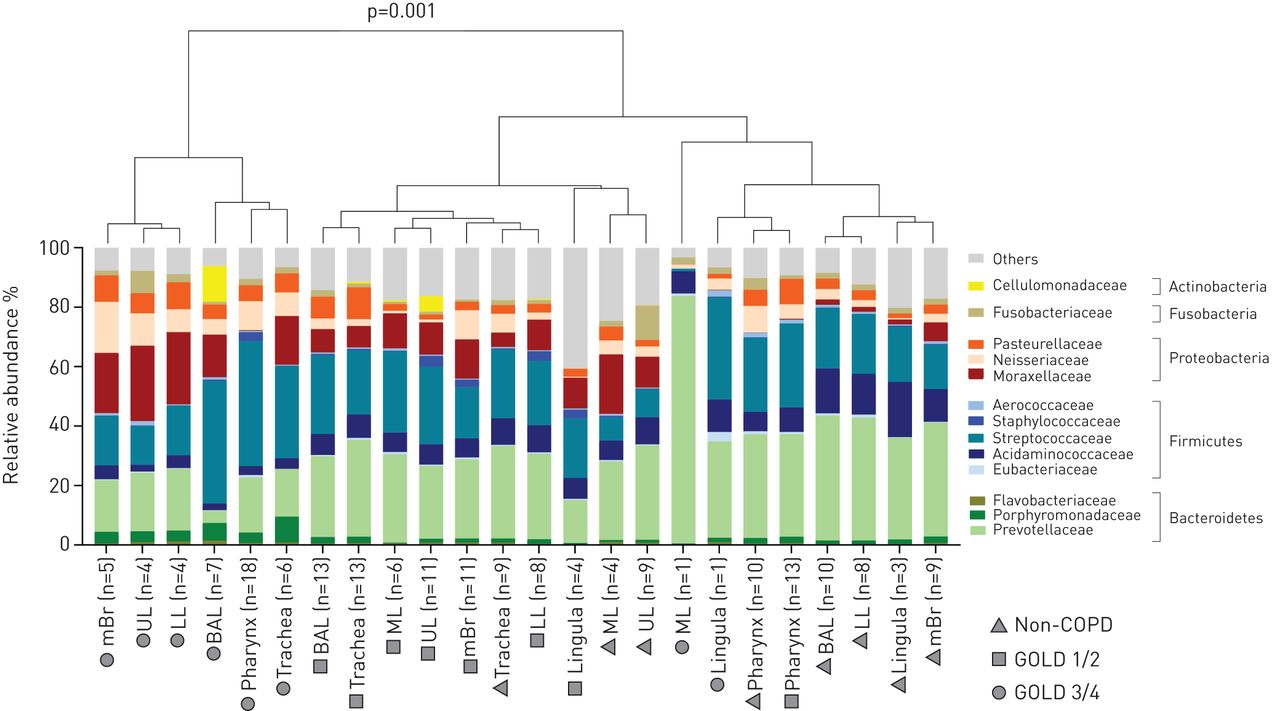
Bacterial community comparison of samples with the most abundant bacterial families (>0.5% mean relative abundance). Illustrated are the samples for the upper lobe (UL), middle lobe (ML), lower lobe (LL), main bronchus (mBr), bronchoalveolar lavage (BAL), lingula, trachea and oropharynx. Samples were grouped according to the site of the respiratory tract and disease status (controls, Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 1/2 and GOLD 3/4). The tree was produced with hierarchical clustering (hclust from the stats package in R using the ward method). Bacterial composition of chronic obstructive pulmonary disease (COPD) GOLD 3/4 patients (mBr, UL, LL, BAL, pharynx and trachea) differed significantly from samples of controls and GOLD 1/2 patients, as measured by permutational multivariate ANOVA (adonis function of the vegan package in R) (p=0.001, stress 0.196).
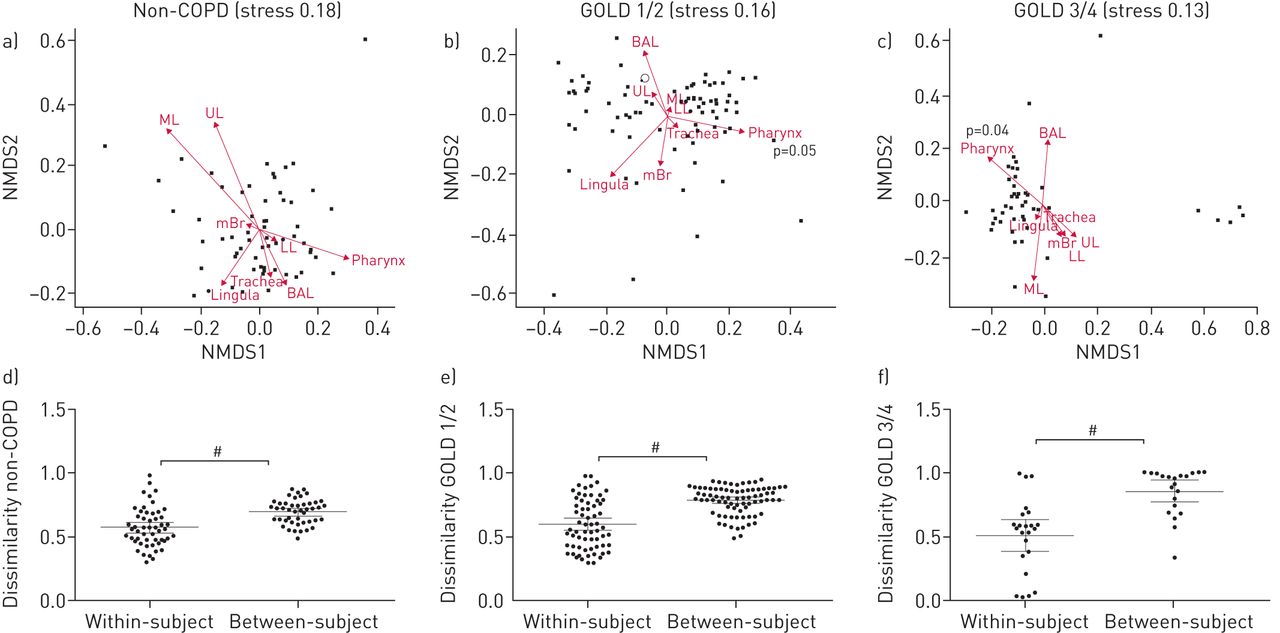
β-diversity analyses between sampling sites. β-diversity was calculated using the abundance-based operational taxonomic unit table as input. a–c) Relative abundance-based Jaccard dissimilarity values were graphically represented using nonmetric multidimensional scaling (NMDS) as ordination method. Stress values are indicated. Arrows indicate clustering of samples according to the sampling site and were fitted using the envfit function of R. UL: upper lobe; ML: middle lobe; LL: lower lobe; mBr: main bronchus; BAL: bronchoalveolar lavage. a) No significant difference in overall microbiota composition between the different sites of controls using permutational multivariate ANOVA (MANOVA). b) Samples from pharynx clustered significantly differently in chronic obstructive pulmonary disease (COPD) Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 1/2 patients (permutational MANOVA, p=0.05). c) Samples from pharynx clustered significantly differently in COPD GOLD 3/4 patients (permutational MANOVA, p=0.04). d–f) Between-subject dissimilarity was measured by comparison of BAL samples from all patients within the corresponding group. Within-subject dissimilarity represents all dissimilarity values of a subject within the corresponding group. Within-subject dissimilarity was significantly lower in all three groups than the between-subject dissimilarity. #: p<0.0001, Mann–Whitney test.
BAL cell gene expression significantly groups GOLD 3/4 patients
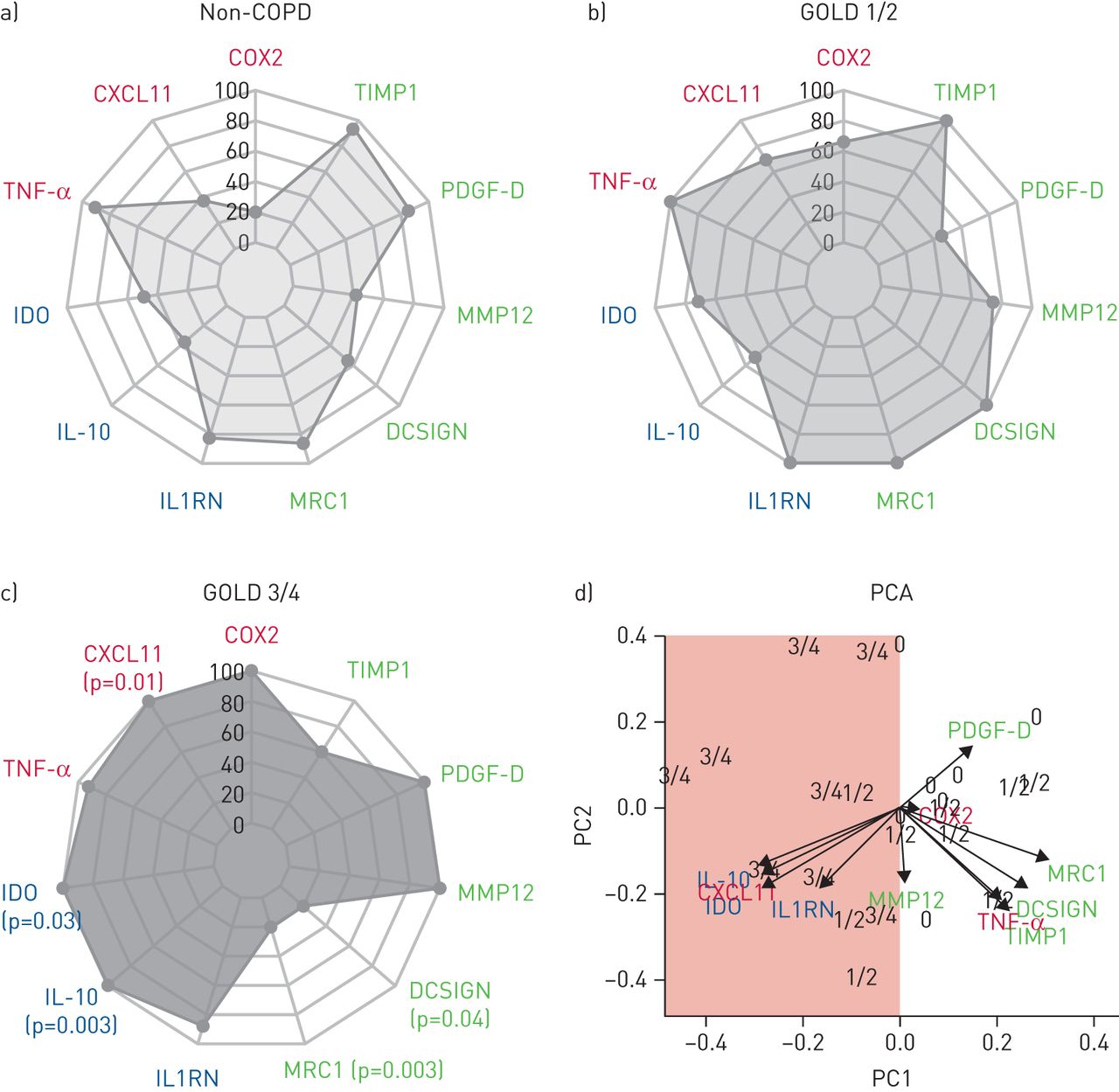
Next, gene expression analyses were performed based on 39 out of 42 BAL samples from COPD patients and controls (three samples had to be excluded because of insufficient RNA yield). As for inflammatory markers, the number of CXCL11 gene transcripts increased with COPD severity degree and was highest in GOLD 3/4 patients (figures 3a–c and S3) (Mann–Whitney test, p=0.01). For immunomodulatory markers, a higher expression of IDO and IL-10 was seen in COPD GOLD 3/4 compared to controls (p=0.03 for IDO; p=0.003 for IL-10). Regarding markers for scavenging receptors and remodelling, significantly reduced gene expression was seen for MRC1 and DCSIGN in COPD GOLD 3/4 patients compared to both controls (p=0.003 for MRC1; p=0.04 for DCSIGN) and GOLD 1/2 patients (p=0.001 for MRC1; p=0.0004 for DCSIGN) (figures 3a–c and S3). Overall, gene expression analyses clearly separated GOLD 3/4 patients from controls and GOLD 1/2 patients, as illustrated in the PCA (figure 3d). The data for GOLD 3/4 within the PCA plot are widely scattered, again illustrating increased heterogeneity of the host response among GOLD 3/4 patients, as compared to the controls and to GOLD 1/2 patients.

Expression of bronchoalveolar lavage cell markers. The relative abundances of inflammatory (red), immunomodulatory (blue) and remodelling/scavenging receptor (green) markers are shown. Results are shown according to a) controls, b) chronic obstructive pulmonary disease (COPD) Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 1/2 patients and c) COPD GOLD 3/4 patients. p-values (Mann–Whitney tests) of GOLD 3/4 patients compared to controls are indicated in (c). In addition, d) principal component analysis (PCA) based on the selected gene targets is shown. Mean values of the three groups were received and the highest mean value of each cell marker was set as 100% while the other values are relative means. A clear separation of GOLD 3/4 patients (3/4) from the controls (0) is illustrated (red surface). Arrows indicate clustering of samples according to the severity of COPD (i.e. controls (0), COPD GOLD 1/2 (1/2) and COPD GOLD 3/4 (3/4)). COX: cyclo-oxygenase; TIMP: tissue inhibitor of metalloproteinase; PDGF: platelet-derived growth factor; MMP: matrix metallopeptidase; DCSIGN: dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin; MRC: mannose receptor C; IL1RN: interleukin-1 receptor antagonist; IL: interleukin; IDO: indoleamine 2,3-dioxygenase; TNF: tumour necrosis factor; PC: principal component.
Correlation of the BAL cell gene expression and the microbiota
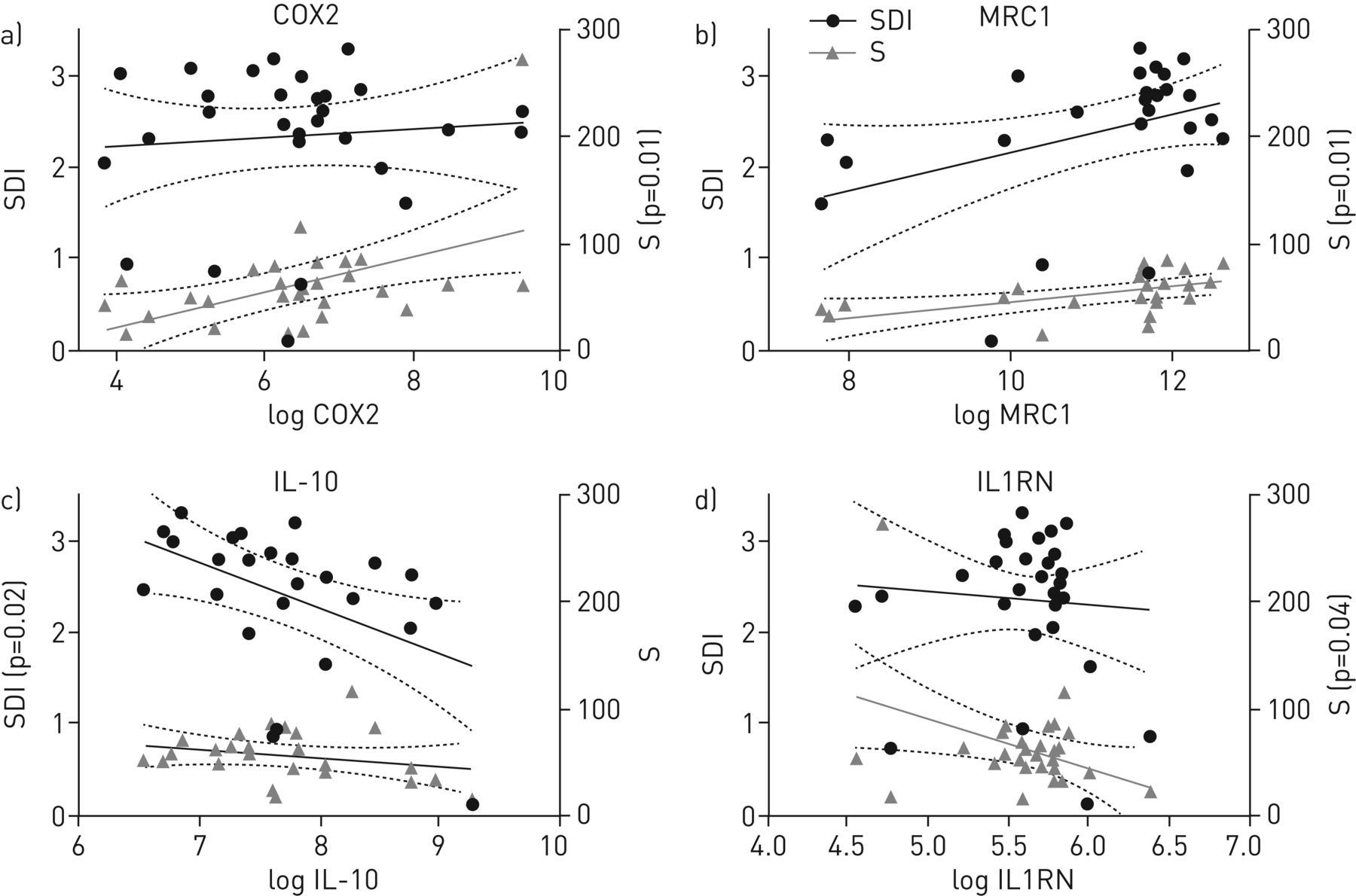
We then performed correlation analysis of BAL cell gene expression results with the paired α-diversity values SDI and richness (figures 4 and S4). These analyses were independent of the COPD severity classifications as defined by the clinicians. Pearson correlation revealed a positive association with richness for COX2 (p=0.01) and MRC1 (p=0.01). In contrast, a negative correlation was found for the SDI and richness for IL-10 (p=0.02) and IL1RN (p=0.04). Employing a correlation analysis based on bacterial families of the microbiota, Moraxellaceae were positively correlated with IL-10 (p=0.04), CXCL11 (p=0.04), IDO (p=0.02) and TNF-α (p=0.001), while the opposite was true for Streptococcaceae and TNF-α (p=0.02) (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation of expression values of bronchoalveolar lavage cell markers with α-diversity measurements: Shannon Diversity Index (SDI) and richness (S). Illustrated are the cell markers with significant correlations with α-diversity, namely a) cyclo-oxygenase (COX)2, b) mannose receptor C (MRC)1, c) interleukin (IL)-10 and d) IL-1 receptor antagonist (IL1RN) (p-values are derived from Pearson correlation). The linear regressions (solid lines) with 95% confidence intervals (dotted lines) are indicated. Only significant p-values are shown (i.e. p<0.05).
Integrative analyses of the BAL cell gene expression and the microbiota
However, relative abundances of bacterial families were not normally distributed and we therefore performed univariate negative binomial regression modelling to investigate associations between the relative abundance of bacterial families and paired data of the BAL cell gene expression of the host (table S2). We found a positive association of Flavobacteriaceae and PDGF-D (figure S5 and table S2) (negative binomial regression model, estimate±se 0.07±0.03, p=0.03).
Discussion
While there is an increasing body of information on the microbiota and host immune responses in COPD exacerbation [19–24], data from stable COPD patients are still scarce. Here, we performed a cross-sectional study of 32 stable COPD patients with different disease severity degrees and compared these to 10 controls. Overall, this is, to the best of our knowledge, the first study investigating the airway microbiota of different sites of the respiratory tract in COPD patients with different disease severity degrees, in combination with host immune gene expression data of BAL cells.
In our study, the microbiota composition of COPD patients was mirrored by a decreased abundance of commensal bacteria, such as the family of Prevotellaceae, which is in line with previous findings from other studies [1–3, 25] and, together with differences in the β-diversity analyses, clearly demonstrated a disordered microbiota in patients with COPD. However, despite the GOLD stratification into different lung functional severity degrees (1–4) and risk classes (A–D), COPD is a highly heterogeneous disease [26, 27]. This is also reflected by our microbial between-subject dissimilarity finding that was most pronounced in more advanced COPD GOLD 3/4 patients and may suggest that the heterogeneity of the microbiota may need to be taken into account when defining COPD phenotypes [27].
A major strength of our study is that we also investigated the differences in the microbiota among the different sampling sites within the respiratory tract. In general, our data indicated a rather homogenous microbiota along the respiratory tract. This finding is consistent with a previous study by Charlson et al. [28] performed in healthy subjects. In addition, we also hypothesise that bacteria enter the lungs in health primarily by microaspiration, as previously suggested [29]. Our results somewhat diverge from a study by Erb-Downward et al. [2] that described a compartmentalisation of the respiratory tract microbiota in COPD. In their study, explanted lung tissue was utilised, which may partly account for the differences observed. Taken together, and excluding the oropharynx with its indeed different profile in COPD patients, we hypothesise a homogenous microbiota along the respiratory tract of stable patients with COPD and controls. This may therefore support a more general approach to treating a disordered microbiota in COPD in the future. However, such a future strategy needs to take into account the heterogeneity of COPD with exceptions for individual patients and circumstances may differ during exacerbations.
A recent study found a correlation between decreased sample diversity in COPD patients and forced expiratory volume in 1 s (FEV1) as a clinical marker of the disease [1]. Consistent with our data and due to the fact that COPD is heterogeneous, this suggests that the sample diversity may be employed as a surrogate marker of the degree of microbiota disordering in COPD, rather than actual bacterial abundances. Within our study, we correlated sample diversity and richness with the host immune responses rather than FEV1, offering an additional insight into the pathogenesis of COPD. Not only did expression of both immunomodulatory IL-10 and scavenging receptor and remodelling marker MRC1 correlate with bacterial diversity or richness, but gene expression was also significantly associated with more advanced COPD (GOLD 3/4) patients. Comparing the immune stimulatory capacity on human monocyte-derived dendritic cells of pathogenic Haemophilus spp. and Moraxella spp. revealed that, similar to our study, the bacteria provoked a higher production of IL-10 compared to other “commensal” bacteria [30]. Regarding the analysis of scavenging receptor and remodelling markers of macrophages, we found significantly decreased expression of MRC1 and a positive correlation with species richness in COPD GOLD 3/4 patients. Madsen et al. [31] demonstrated in a mouse model that a genetic knockout of the mannose receptor MRC1 led to impaired intracellular collagen degradation. Collagen degradation is required for tissue remodelling and repair [31], and disturbed collagen homeostasis might thus indicate a perturbed homeostasis of tissue repair and degradation in COPD patients. Our study expanded investigations beyond establishing a mere library of pulmonary microbiota, and provides evidence for significant correlations between the microbiota and the host immune response. A recent study suggested that the host immune response to microorganisms in the lung microbiota contributed to the pathogenesis of GOLD 4 COPD patients [32], and this link was further mechanistically investigated in a COPD mouse model [33]. Though we now provide the most comprehensive study in the field, together with the other two studies, we were not able to answer the question of whether the altered gene expression drives the disordering of the microbiota in COPD or vice versa.
There are limitations to our study. Even though we included a reasonably high number of samples from COPD patients, the study size still remains a limiting factor for a microbiota study due to the observed heterogeneity in patients. Based on accurate power analyses [34], more individuals should be included, although it is challenging to find sufficient volunteers allowing brushing of the respiratory tract at different sites. However, protected sampling is preferable in bronchoscopic studies of the airway microbiome to avoid contamination [35]. Furthermore, the groups we compared received different COPD medication, which has been shown to influence the microbiota composition [4]. Especially for severely and very severely diseased COPD patients, we found a high between-subject dissimilarity, which indicates a heterogeneous microbiota composition between the different patients. It is therefore difficult to unravel the effects of different covariates and the disease itself on the microbiota composition. In order to adjust for covariates, larger longitudinal studies will be required in the future. A further limitation of our study is the inclusion of two controls that had received long-acting β2-agonists (LABA) plus inhaled corticosteroids (ICS) for symptomatic treatment of cough, but without evidence for a respiratory condition. Within this study, the inclusion of completely healthy controls was challenging due to ethical issues, but neither the smoking controls nor the participants with LABA+ICS revealed a disordered microbiota as compared to all the other controls. Finally, since BAL cells were not sorted prior to total RNA isolation, the results pertain to total BAL cells. Although the set of gene targets that were selected are primarily transcribed in macrophages, a partial contribution of other cells, especially neutrophils, cannot be excluded.
In conclusion, we showed that the respiratory tract microbiota in COPD patients is disordered when compared to controls, and is characterised by a lower diversity and a different bacterial composition. The disordered microbiota is closely associated with an altered macrophage gene expression profile in BAL cells. These data provide an extensive description of host and microbiota factors in different COPD severity degrees, and may thus contribute to an improved understanding of factors determining the course and prognosis of disease in COPD patients.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00015-2018_supp
Supplementary figures 00015-2018_supp_figs
Acknowledgements
We are most grateful to all study participants and the team at the Bronchoscopy Unit of the Inselspital Bern for their assistance. We thank Suzanne Aebi (Institute for Infectious Diseases, University of Bern, Bern, Switzerland) for her laboratory assistance, and Liselotte McEvoy and Sandra Barnowski (Dept of Clinical Research, University of Bern), as well as Irène Stutz, Susanna Glaus and Therese Gerber (Institute for Infectious Diseases) for their assistance in data collection.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Conflict of interest: None declared.
Support statement: We gratefully acknowledge funding of this study by a Swiss National Science Foundation Sinergia grant (CRSII3–141875) to B.J. Marsland, C. von Garnier and M. Hilty. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received January 29, 2018.
- Accepted April 28, 2018.
- Copyright ©ERS 2018
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










