





Abstract
Background Combined pulmonary fibrosis and emphysema (CPFE) has recently received great attention, with studies suggesting that it presents a distinct clinical entity while others have challenged this hypothesis. This nationwide study aimed to describe a large cohort of Greek CPFE patients and to examine potential prognostic factors for survival.
Methods This retrospective study included 97 patients with CPFE. Demographic and clinical data, pulmonary function tests, echocardiography results and bronchoalveolar lavage analysis were recorded.
Results Most patients were male (94.8%) and 92% were current or ex-smokers. Spirometry results were abnormal (forced vital capacity (FVC) 72.9±19.9% pred and forced expiratory volume in 1 s/FVC 82.9±9.7%) with reduced diffusing capacity of the lung for carbon monoxide (DLCO) (42.3±17.4% pred). Mean systolic pulmonary arterial pressure was 41.9±19.7 mmHg and pulmonary hypertension was present in 58.8% of patients. Mean 6-min walk distance was 335.4±159.4 m. Mean emphysema score was 14.23±8.69% and mean interstitial lung disease (ILD) extent was 39.58±19.82%. Mean survival was 84 months (95% CI 72–96 months). Patients with DLCO ≥39% pred had better survival than patients with DLCO <39% pred (p=0.031). Patients with ILD extent ≥30% had worse survival than patients with ILD extent <30% (p=0.037).
Conclusions Our results indicate that CPFE patients have preserved lung volumes associated with disproportionately reduced DLCO, while reduced DLCO and increased ILD extent was associated with worse prognosis.
Abstract
Prognosis of CPFE is associated with pulmonary function status and ILD extent http://ow.ly/izvd30nHFgh
Introduction
Combined emphysema with pulmonary fibrosis (CPFE) has recently gathered increasing interest, and researchers have proposed that CPFE may represent a distinct clinical syndrome with specific clinical, radiological and functional characteristics, along with a different prognosis [1, 2]. According to previous studies, CPFE patients have preserved lung volumes with greatly reduced diffusing capacity of the lung for carbon monoxide (DLCO). Clinicians have suggested that CPFE is a distinct clinical syndrome, while the recently published guidelines concerning the classification of idiopathic interstitial pneumonias have not included CPFE as an individual entity but have described it as a coexisting pattern of emphysema with chronic fibrosing interstitial pneumonias, underlining the need for further investigations [3]. Additionally, CPFE is associated with a high prevalence of pulmonary hypertension [4]. In terms of prognosis, the data in the literature are rather contradictory. Some have reported worse outcome in CPFE patients [5] while others have observed better prognosis when compared to idiopathic pulmonary fibrosis (IPF) without emphysema [6].
The objective of the present study was to provide a detailed description of the characteristics of CPFE in a Greek cohort and to determine factors that may influence survival.
Methods
Study design
This retrospective, multicentre study was conducted by the Hellenic Interstitial Lung Diseases Group. Patients presenting to Greek referral centres and evaluated by physicians with a special interest in interstitial lung diseases (ILDs) were included in this study between March 2011 and December 2014. Patients were recruited by seven Greek ILD centres, namely the Respiratory Medicine Dept of the University of Thessaly (University Hospital of Larissa; 16 patients), the Dept of Pneumonology of the University of Thessaloniki (G. Papanikolaou Hospital; 13 patients), the Dept of Thoracic Medicine and Interstitial Lung Disease Unit of the University Hospital of Heraklion (18 patients), the 1st Dept of Pneumonology of “SOTIRIA” Athens Chest Hospital (National and Kapodistrian University of Athens; 27 patients), the Respiratory Medicine Dept of the “Sismanoglion” General District Hospital (Athens; five patients) and the Dept of Pneumonology of the University Hospital of Alexandroupolis (18 patients).
Only patients with a chest high-resolution computed tomography (HRCT) scan available for review were included in the study. The study was approved by the respective ethics committee of each institution. All patients included provided written and verbal consent. Some of these patients were included in a previous study [7].
Study population
Subjects were included in the study if the following criteria were met: 1) consistent clinical presentation; 2) presence of emphysema on HRCT scan defined as well-demarcated areas of decreased attenuation with very thin wall (<1 mm) or no wall in the upper zones of the lungs; and 3) presence of HRCT features consistent with definite usual interstitial pneumonia (UIP) such as reticular opacities, honeycombing and/or traction bronchiectasis with basal and subpleural predominance, and absence of features inconsistent with UIP [8]. In a subgroup of patients who did not fulfil the HRCT criteria for definite UIP, the diagnosis was established by surgical lung biopsy. We chose to include only patients with a firm diagnosis of IPF in order to avoid bias due to inclusion of patients with other type of ILDs with different prognosis and, most of the time, better prognosis than IPF. Demographic and clinical data were obtained from all participants, including symptoms, smoking status, pulmonary function tests (PFTs), 6-min walk test results, arterial blood gas analysis, serology for autoantibodies and bronchoalveolar lavage fluid analysis. The use of right heart catherization, which is the gold standard for the diagnosis of pulmonary hypertension for research purposes alone, is associated with poor patient consent and, therefore, the presence of pulmonary hypertension was assessed by transthoracic echocardiogram (which presents a noninvasive and easily applicable method). Pulmonary hypertension was defined as systolic pulmonary arterial pressure (sPAP) ≥35 mmHg [9–11]. sPAP was estimated as previously reported [11]. Linear internal measurements of the left ventricle and its walls were performed in the parasternal long-axis view while right ventricle dimensions were estimated from a right ventricle-focused apical four-chamber view.
Survival time was assessed as follows: for patients that died during the study period, survival time was calculated from the date of diagnosis to the date of death; for patients that were alive at the end of the study period, survival time was calculated from the date of diagnosis to the date of the end of the study (i.e. December 2014).
Radiological features
All patients had a HRCT of the chest performed and reviewed by two thoracic radiologists (K. Malagari and A. Oikonomou) who were blinded to clinical and histopathological data. The following features were quantified at five levels as previously described [12] (apex to origin of great vessels, main carina, pulmonary venous confluence, between the third and fifth level, and 1 cm above the hemidiaphragm). The overall extent of ILD (reticular pattern and ground-glass opacification) was estimated to the nearest 5%. The extent of emphysema, defined as well-demarcated areas of low attenuation emarginated by thin wall (<1 mm) or no wall, was quantified to the nearest 5%. For a more comprehensive analysis of quantification of ILD and emphysema extent, the reader is referred to the supplementary material.
Statistical analysis
Data are presented as mean±sd unless otherwise indicated. Normal distribution was assessed by the Kolmogorov–Smirnov test. A survival curve was determined using the Kaplan–Meier method and was analysed for statistical significance using the log-rank test. Statistical analysis was performed using the SPSS 16 statistical package (SPSS Inc., Chicago, IL, USA). Statistical significance was defined as p<0.05.
Results
Demographic and clinical characteristics of the participants are shown in table 1. The study population consisted of 97 subjects with a mean±sd age of 68.2±9.0 years. Of the patients studied, 94.8% were male. ∼92% of patients were current or ex-smokers while the remaining were lifelong nonsmokers. As far as comorbidities is concerned, 14.43% of patients presented coronary artery disease, 58.76% of patients presented arterial hypertension and 25.97% of patients presented diabetes mellitus. Histological confirmation of the UIP pattern was available in 26.8% of subjects. Finger clubbing was documented in 63.9% patients. All patients had dyspnoea and 75.2% of subjects reported cough. The mean disease duration (estimated from diagnosis until the inclusion of the patient in the study) was 3.5±2.8 years. Increased autoantibodies titres (antinuclear antibodies and/or rheumatoid factor) were present at 9.27% of the study population.
Demographic characteristics and clinical manifestations of the study population
The results of the PFT analysis are shown in table 2. Mean values from spirometry indicated a mild restrictive ventilatory defect. Gas exchange was severely impaired as indicated by the reduced DLCO. Lung volumes were moderately reduced (table 2). Mean arterial oxygen tension at rest was reduced and as expected, mean alveolar–arterial oxygen tension gradient was increased (table 2). Data from the transthoracic echocardiogram were available in 68 patients. Mean sPAP was 41.9±19.7 mmHg and pulmonary hypertension was present in 58.8% of the study participants. Mean left ventricle ejection fraction was 62.02±9.44%, mean left ventricular end-systolic dimension was 32.80±7.83 mm, mean left ventricular end-diastolic dimension was 36.23±7.89 mm and mean right ventricle basal dimeter was 36.57±6.45 mm. Mean 6-min walk distance was 335.4±159.4 m.
Pulmonary function tests, arterial blood gas analysis and systolic pulmonary arterial pressure (sPAP) data of the study population
Bronchoalveolar lavage fluid analysis was available in 51.5% of the participants. The mean total cell count was 17.7±14.0×105 per mL. The differential cell counts are shown in table 3. Mean emphysema score was 14.2±8.6% and mean ILD extent was 39.6±19.8%.
Bronchoalveolar lavage (BAL) fluid analysis and high-resolution computed tomography (HRCT) findings
Survival analysis
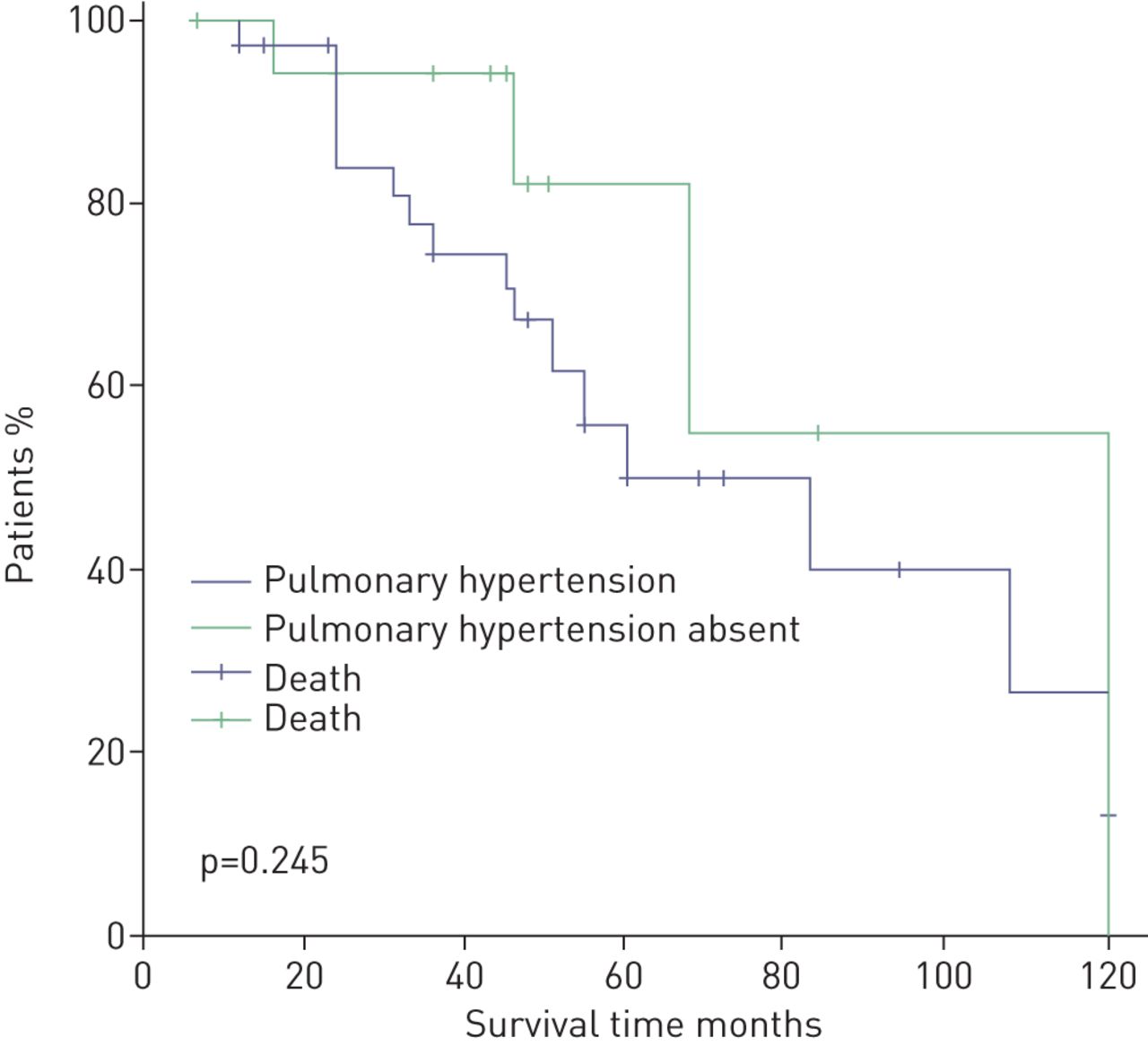
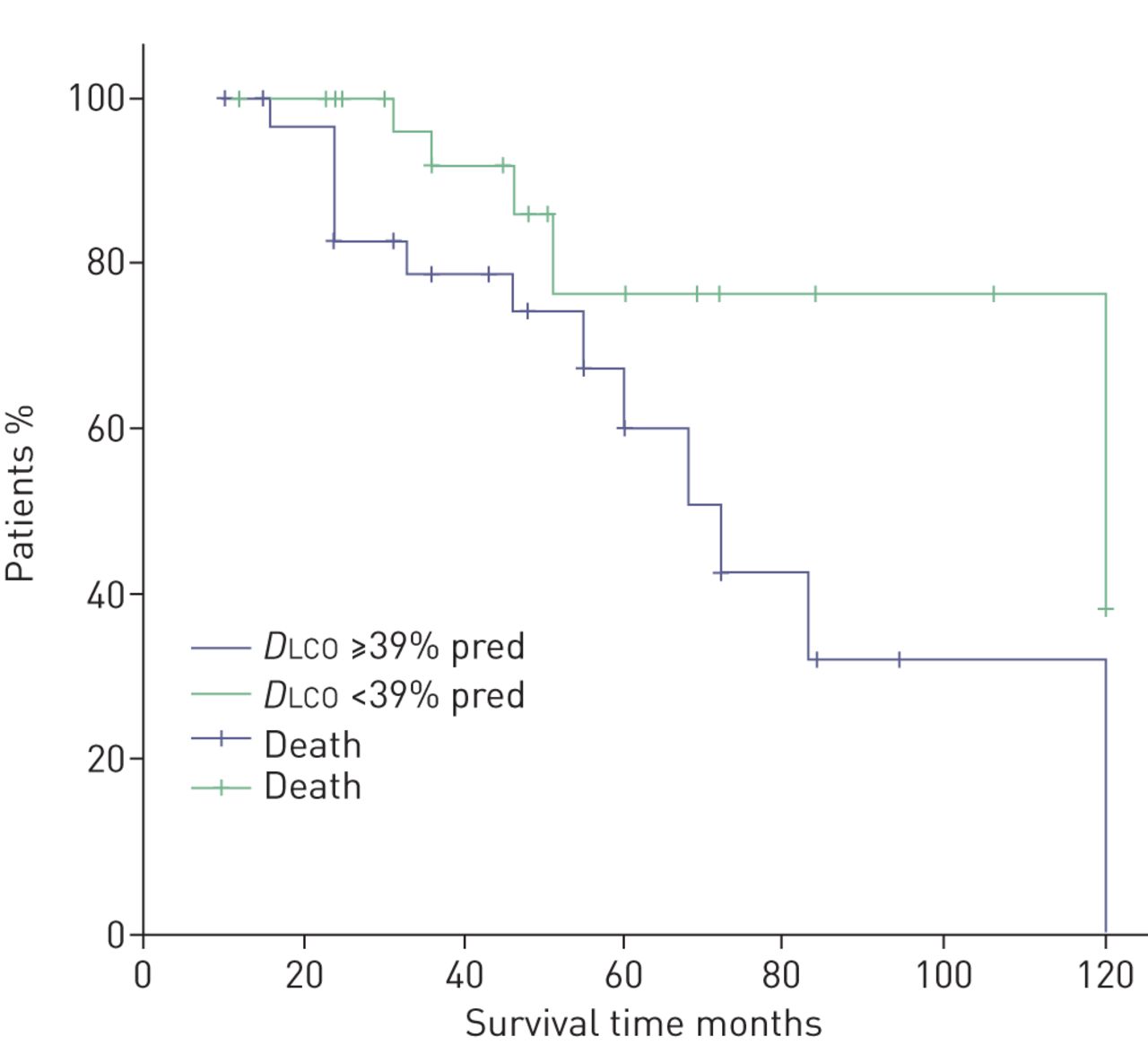
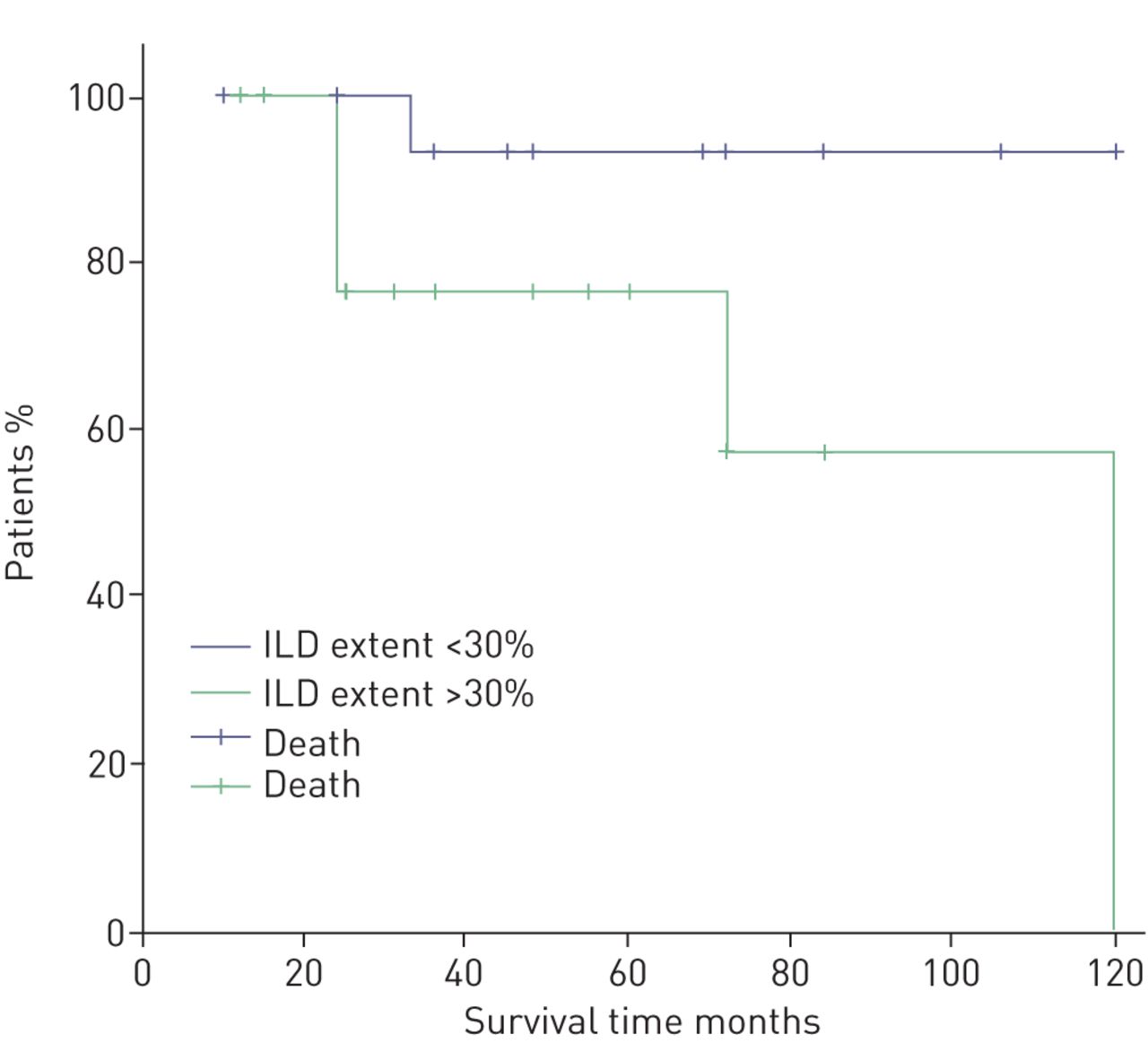
Mean survival was 84 months (95% CI 72–96 months) after the diagnosis. We stratified the patient group according to the presence or absence of pulmonary hypertension as assessed by Doppler echocardiography using the threshold of sPAP 35 mmHg. We observed that patients with sPAP ≥35 mmHg did not differ in terms of prognosis with patients with sPAP <35 mmHg (figure 1). We additionally stratified patients according to values of sPAP (i.e. sPAP <30, 31–40, 41–50 and >51 mmHg) and did not observe differences in prognosis (data not shown). Additionally, we stratified patients according to disease severity (as assessed by DLCO % pred) as mild to moderate (DLCO ≥39% pred) or severe (DLCO <39% pred). We observed that patients with DLCO ≥39% pred had better survival than patients with severe disease (p=0.031) (figure 2). Interestingly, patients with ILD extent ≥30% presented worse prognosis than those with ILD extent <30% (p=0.037) (figure 3). The presence of comorbidities (i.e. coronary artery disease, hypertension or diabetes mellitus) or therapy status were not determinants of prognosis (data not shown).

Kaplan–Meier curve of combined pulmonary fibrosis and emphysema patients stratified by the presence or absence of pulmonary hypertension as assessed by echocardiography (i.e. systolic pulmonary arterial pressure ≥35 mmHg).

Kaplan–Meier curve of combined pulmonary fibrosis and emphysema patients stratified by diffusing capacity of the lung for carbon monoxide (DLCO) % pred.

{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curve of combined pulmonary fibrosis and emphysema patients stratified by interstitial lung disease (ILD) extent.
Discussion
In this multicentre study, we observed that CPFE subjects presented a mild restrictive spirometry associated with reduced gas exchange. Moreover, reduced DLCO and increased ILD extent were associated with poor prognosis. Mean survival in our population was 84 months. Notably, the majority of our patients presented signs of vascular involvement as sPAP was 41.92 mmHg. In this respect, our observations give further insight in the clinical course of the CPFE population.
We observed that CPFE patients in our cohort presented a rather subnormal spirometry with reduced DLCO possibly due to the additive effects of the two diseases in DLCO and their opposing effects in elastic recoil [4]. Our results are in accordance with previously published data indicating that spirometry values, and mainly FVC alone, cannot be used in order to screen or to monitor patients with CPFE and do not allow an accurate assessment of prognosis. Additionally, we observed that static lung volumes were reduced with a mean total lung capacity of 66.43±16.80% pred, indicating a predominantly restrictive pattern. Previous published data have reported normal static lung volumes in CPFE patients [13], while others have demonstrated a mild reduction. Todd et al. [6] have reported that lung volumes vary according to the extent of emphysema and thus may explain the discrepancy among the studies.
The natural history of CPFE is largely unknown. We show that the mean survival in our cohort was 84 months following diagnosis. We chose to include only patients with an IPF pattern in order to avoid bias due to inclusion of patients with other type sof ILDs with different prognoses to IPF. We did not include a control group of IPF subjects; therefore, we cannot directly compare the CPFE survival with that of IPF. However, according to previously published data, it seems that CPFE subjects carry equal or even better prognosis versus IPF alone [6, 13–16] while others have challenged this conclusion [17, 18]. The reason(s) for these differences in survival of CPFE compared to IPF are not clear but one could argue that this may be the result of the overlapping pathophysiological mechanisms implicated in the two entities, resulting in different clinical course of IPF or emphysema alone [18, 19]. Another possible explanation could be differences in the prevalence of comorbidities such as lung cancer [20]. Differences in metalloproteinase expression [21], surfactant protein C gene mutations [22], overexpression of tumour necrosis factor-α [23] and platelet-derived growth factor-B [24] in animal models have been implicated, among other factors, in the development of CPFE. Additionally, the discordance among the studies concerning CPFE versus IPF survival may be due to different definitions of CPFE among studies. The present study was not designed to address this hypothesis that needs further investigation in future studies.
Studies have previously shown that decline in DLCO is associated with worse prognosis in CPFE subjects [25]. Our results are in accordance with previous findings and indicate that DLCO may serve as a predictor of mortality in the CPFE population. We demonstrated that DLCO <39% pred is associated with worse prognosis when compared to patients with DLCO ≥39% pred. Some have suggested that DLCO may correlate with the degree of severity and parenchymal destruction better than other physiological parameters like FVC due to the opposing effects of the two diseases in elastic recoil [26, 27]. However, it should be stressed that DLCO may be influenced by the presence of pulmonary vascular involvement. Interestingly, others have suggested that a decline in forced expiratory volume in 1 s may be the best marker for predicting mortality in CPFE subjects while DLCO may not accurately identify patients at risk [26].
The association of CPFE with pulmonary hypertension has received significant attention in recent years. Pulmonary hypertension is highly prevalent in CPFE patients and in agreement with earlier studies [2, 4], we demonstrated that >50% of our patient population probably presented pulmonary hypertension, with a mean sPAP of 41.9±19.7 mmHg. The presence of pulmonary hypertension has previously been recognised as a negative predictor of mortality in CPFE patients. However, we did not observe any differences in survival in patients with and without pulmonary hypertension. One possible explanation for the discrepancy is the use of transthoracic echocardiography to diagnose pulmonary hypertension and/or the lack of a consensus definition of CPFE in the literature. We have included only subjects with a UIP pattern either in HRCT or lung tissue biopsy, while others have used less strict criteria, thus including patients with a nonspecific interstitial pneumonia pattern in the analyses [28].
In the present study, we observed that increased ILD extent may have a negative impact on prognosis. Others have challenged this finding by reporting no influence of pulmonary fibrosis score on survival [18] while Jacob et al. [16] reported that the total extent of fibrosis and emphysema predicts mortality in CPFE patients. Our observations suggest that CPFE outcome may be worsened by the IPF component of the disease. ILD extent may reflect overall severity of lung parenchyma involvement and through that mechanism, may be implicated in CPFE survival. Additionally, we did not observe any association of therapy status with survival. At the time the study was performed, the only available therapy in Greece was the triple combination of azathioprine, prednisolone and N-acetylcysteine. The small number of patients receiving therapy in our cohort renders major uncertainty to the latter finding.
Our study has several limitations. One major criticism of our findings is the lack of right heart catheterisation as a more accurate measure of pulmonary haemodynamics and as a means of assessing the presence of pulmonary hypertension, which may be implicated in the diversity of our results with those previously published [4]. We acknowledge that Doppler echocardiography may be misleading in the assessment of patients with suspected pulmonary hypertension and may not adequately identify pulmonary hypertension, especially in the setting of lung disease [29]. Right heart catheterisation, which is a rather invasive method, is the gold standard for the diagnosis of pulmonary hypertension. However, echocardiography serves as a noninvasive and easily applicable method that is better tolerated by the patient for research purposes alone and, therefore, is associated with better patient consent. Taking this into account, we acknowledge that based on our findings, no definite conclusions can be drawn concerning the prevalence and prognostic ability of pulmonary hypertension in patients with CPFE. Additionally, our study has an observational and retrospective design, and therefore our patients may be subject to selection bias. Another limitation is the lack of serial PFT data that might provide a helpful predictor of prognosis. Finally, we acknowledge that to have a complete evaluation of survival analysis, patients with CPFE need to be compared with a matched group of IPF patients and a second group of emphysema patients.
Conclusions
In conclusion, we have observed that patients with CPFE present preserved lung volumes associated with disproportionately reduced DLCO. Reduced DLCO and increased ILD extent was associated with worse prognosis. Further clarifications are warranted to clarify the clinical course and the pathophysiological mechanisms involved in CPFE.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Quantification of emphysema and fibrosis 00014-2018.supplementary_material
Acknowledgements
We acknowledge the contribution of the following colleagues, who made this report possible. K. Manika (Aristotle University of Thessaloniki G. Papanikolaou Hospital, Thessaloniki, Greece), F. Bardaka (University of Thessaly, Larissa, Greece), G.A. Margaritopoulos (University Hospital of Heraklion, Heraklion, Greece), A. Tzouvelekis (“SOTIRIA” Athens Chest Hospital, 1st Dept of Pneumonology, Athens, Greece) and F. Drakopanagiotakis (“Sismanoglion” General District Hospital, Athens, Greece).
Footnotes
This article has supplementary material available from openres.ersjournals.com.
Conflict of interest: F. Malli has nothing to disclose.
Conflict of interest: D. Papakosta has nothing to disclose.
Conflict of interest: K. Antoniou has nothing to disclose.
Conflict of interest: M. Dimadi has nothing to disclose.
Conflict of interest: V. Polychronopoulos has nothing to disclose.
Conflict of interest: K. Malagari has nothing to disclose.
Conflict of interest: A. Oikonomou has nothing to disclose.
Conflict of interest: D.E. Bouros has nothing to disclose.
Conflict of interest: Z. Daniil has nothing to disclose.
- Received January 29, 2018.
- Accepted January 25, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










