





Abstract
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator gene (CFTR) that result in diminished quantity and/or function of the CFTR anion channel. F508del-CFTR, the most common CF-causing mutation (found in ∼90% of patients), causes severe processing and trafficking defects, resulting in decreased CFTR quantity and function. CFTR modulators are medications that increase the amount of mature CFTR protein (correctors) or enhance channel function (potentiators) at the cell surface.
Combinations of CFTR correctors and potentiators (i.e. lumacaftor/ivacaftor, tezacaftor/ivacaftor) have demonstrated clinical benefit in subsets of patients. However, none are approved for patients with CF heterozygous for F508del-CFTR and a minimal function mutation, i.e. a mutation that produces either no protein or protein that is unresponsive to currently approved CFTR modulators. Next-generation CFTR correctors VX-659 and VX-445, each in triple combination with tezacaftor and ivacaftor, improve CFTR processing, trafficking and function in vitro and have demonstrated clinical improvements in phase 2 studies in patients with CF with one or two F508del-CFTR alleles.
Here, we present the rationale and design of four randomised phase 3 studies, and their open-label extensions, evaluating VX-659 (ECLIPSE) or VX-445 (AURORA) plus tezacaftor and ivacaftor in patients with one or two F508del-CFTR alleles.
Abstract
An efficient development programme for VX-659 and VX-445 in triple combination with tezacaftor/ivacaftor in patients with CF with one or two F508del-CFTR alleles was designed based on extensive CFTR modulator experience and pre-clinical and clinical data. http://bit.ly/2UBh6EV
Introduction
Cystic fibrosis (CF) is a rare, severe, multisystem disease affecting more than 80 000 people worldwide [1]. CF is caused by mutations in the CF transmembrane conductance regulator gene (CFTR) that lead to defects in the CFTR protein [2]. CFTR is an ion channel that regulates the transport of chloride and bicarbonate at the cell surface [2–4]. Mutations in CFTR disrupt the activity of the protein by reducing its quantity or function [5]. Clinical manifestations of CF include chronic endobronchial infections, decreased lung function, malnutrition, exocrine pancreatic insufficiency, CF-related diabetes and reduced life expectancy [6]. Approximately 90% of patients with CF carry one or more copies of the F508del-CFTR mutant allele [7], which results in decreased quantity and function of CFTR protein at the cell surface [8].
Based on an understanding of the molecular defects caused by the F508del-CFTR mutation, CFTR modulators that increase the quantity and/or enhance the function of CFTR protein at the cell surface were developed [9–13]. The history of CFTR modulator development was recently reviewed [5]. The systemic delivery of CFTR modulators translates into clinical improvements in multiorgan CF manifestations, including lung function, nutritional status, pulmonary exacerbations and quality of life [14–17]. The first available modulator was the CFTR potentiator ivacaftor (IVA), which augments anion transport by increasing CFTR channel-open probability [9]. IVA monotherapy is approved in selected regions for the treatment of patients with CF with CFTR mutations that produce cell surface-localised CFTR that is responsive to the potentiator, including gating mutations and mutations with residual CFTR function [18–20]. Lumacaftor (LUM) and tezacaftor (TEZ) are CFTR correctors that augment intracellular F508del-CFTR processing and trafficking, thereby increasing the amount of mature CFTR at the cell surface [10–12]. LUM and TEZ, each in combination with IVA, have shown clinical benefit and are indicated in selected regions in patients with CF homozygous for F508del-CFTR (F508del/F508del); TEZ/IVA therapy is also indicated in selected regions in patients with CF heterozygous for F508del-CFTR and a CFTR mutation that is responsive to TEZ/IVA, based on in vitro and/or clinical data [21–24].
Despite the availability of CFTR modulator therapies for many patients with CF, there are no approved CFTR modulators to treat the estimated 30% of patients with CF heterozygous for F508del-CFTR and a minimal function (MF) mutation, defined as a mutation that does not produce protein (e.g. the CFTR nonsense mutation G542X) or produces protein that is unresponsive to TEZ, IVA and the combination of TEZ/IVA (e.g. the CFTR missense mutation N1303K, which results in a severe processing defect) [1, 25]. Furthermore, although TEZ/IVA and LUM/IVA are efficacious in individuals with the F508del/F508del genotype, the magnitude of clinical benefit is not as marked as that experienced by patients with gating mutations treated with IVA, the benchmark of highly effective CFTR modulation [14–16, 26]. We therefore built on the molecular understanding of F508del-CFTR to develop next-generation CFTR correctors that restore high levels of F508del-CFTR activity when used in triple combination with TEZ/IVA in patients with F508del/MF genotypes, who have a single responsive F508del-CFTR allele; the strategy is also intended to maximise CFTR activity in patients with the F508del/F508del genotype. Achievement of these goals was successfully demonstrated in phase 2 trials testing two distinct triple-combination regimens (VX-659/TEZ/IVA and VX-445/TEZ/IVA) in patients with F508del/F508del and F508del/MF genotypes [25, 27]. The studies described herein are designed to validate these early clinical results in a larger group of patients with CF and can potentially bring a next-generation corrector in triple combination with TEZ/IVA to patients with CF with one or two F508del-CFTR alleles, representing significant clinical progress in up to 90% of the population of patients with CF.
Rationale
The F508del-CFTR mutation exhibits multiple molecular defects, including defective cell processing, reduced stability at the cell surface and diminished channel gating [28]. Single-drug correction of cellular misprocessing such as occurs with F508del-CFTR is particularly challenging because multiple intracellular checkpoints exist to remove unstable proteins [5]. VX-659 and VX-445 are next-generation correctors with a different structure and mechanism of action than the first-generation correctors TEZ and LUM [25, 27, 29]. In vitro work in human bronchial epithelial (HBE) cells isolated from patients with CF with F508del/MF or F508del/F508del genotypes demonstrated that a combination of correctors with different mechanisms of action (e.g. TEZ and VX-659 or VX-445) can work additively through complementary mechanisms of action to increase the amount of F508del-CFTR at the cell surface to a higher level than any single corrector [25, 27, 29]. Additionally, triple-combination regimens with next-generation correctors increased in vitro CFTR function to a greater degree than did any dual combination of CFTR modulators [25, 27]. The level of CFTR modulation achieved by these triple-combination treatments in HBE cells with one or two F508del-CFTR alleles met or exceeded the high standard set by IVA in HBE cells expressing G551D-CFTR (∼50% of wild-type CFTR function) [9, 25, 27]. Importantly, the in vitro results observed following IVA treatment in HBE cells expressing G551D-CFTR translated into short- and long-term improvements in clinical outcome in patients with G551D-CFTR mutations treated with IVA (i.e. significant improvements in lung function, nutritional status and patient-reported outcomes), consistent with modification of the underlying disease [9, 14, 30–33]. The pre-clinical and clinical experience with IVA suggests that triple combinations can potentially provide similar levels of clinical efficacy for patients homozygous for F508del-CFTR or heterozygous for F508del-CFTR and an MF mutation [29].
Using an approach unprecedented in scope and breadth in CF clinical research, four novel next-generation correctors (VX-152, VX-440, VX-445 and VX-659) were concurrently evaluated in triple combination with TEZ/IVA in phase 1 and 2 studies in patients with F508del/MF or F508del/F508del genotypes to assess pharmacokinetics, safety and early evidence of efficacy [25, 29]. Four next-generation correctors were evaluated because there is an important medical need, the potential to realise benefit based on prior in vitro and clinical experience was strong, and the inherent risk of drug development is high (including the potential to identify unanticipated toxicity) [25]. TEZ/IVA was chosen as the backbone for the triple-combination regimens because of favourable pharmacological properties, including lack of cytochrome P450 3A induction [16, 17, 34]. To further mitigate risk, initial studies of triple combinations included selected short-term testing in patients with CF after initial safety and pharmacokinetic testing in healthy volunteers (EudraCT trials 2016-003048-35 and 2017-000797-11; ClinicalTrials.gov identifiers NCT03029455 and NCT03227471).
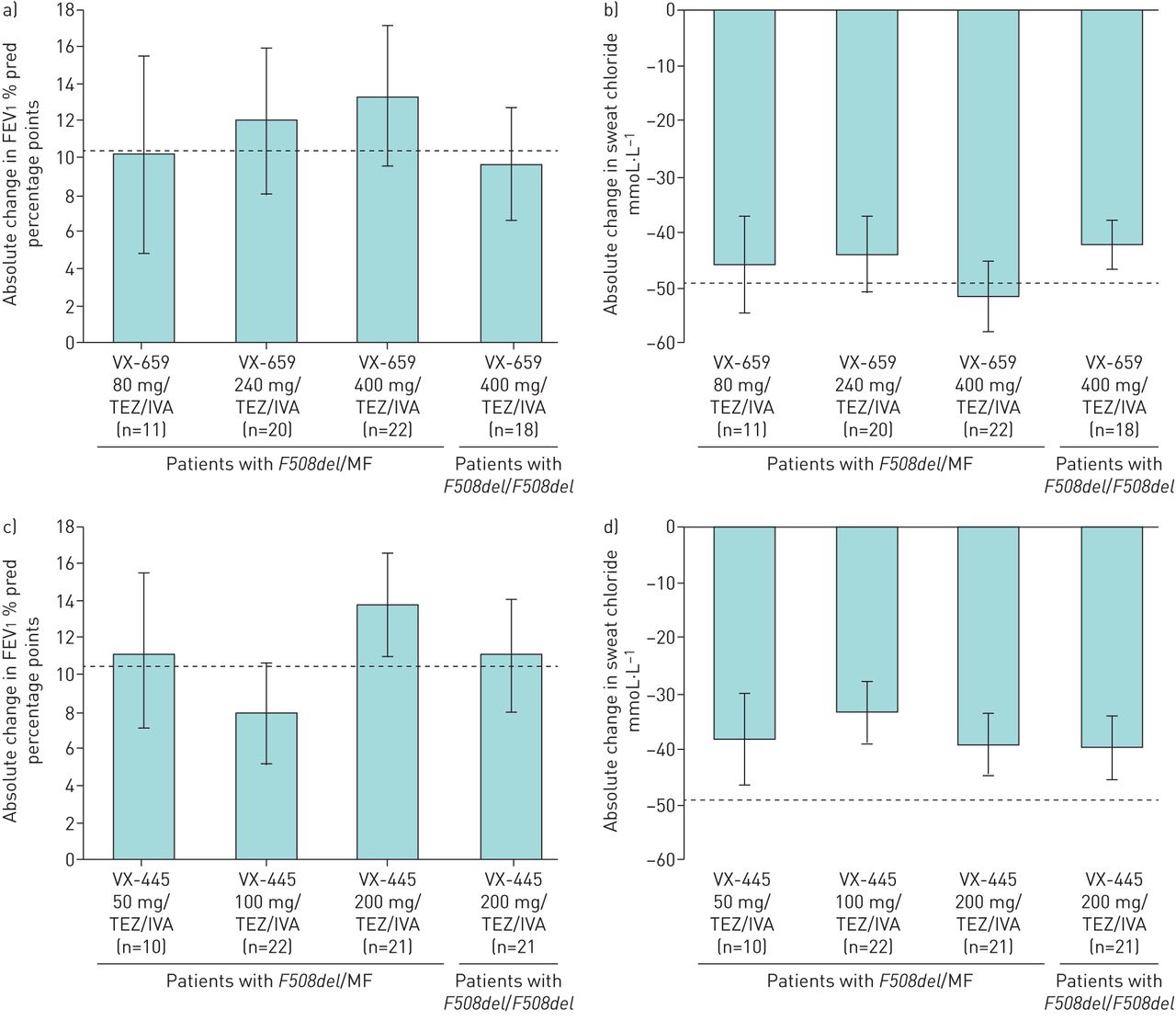
Although all four triple combinations demonstrated preliminary evidence of clinical benefit [25, 27, 35] that was relatively similar in magnitude, two next-generation compounds (VX-659 and VX-445) showed high levels of efficacy together with safety profiles and pharmacological properties suitable for long-term use, and they were selected for further development in phase 3 programmes. VX-659 and VX-445 in triple combination with TEZ/IVA were evaluated in phase 2, randomised, controlled studies in patients with F508del/F508del and F508del/MF genotypes. Different comparator arms were required for each group. For patients with the F508del/F508del genotype, the approved CFTR modulator TEZ/IVA was the comparator and the baseline (following a 4-week run-in period) for within-group changes. The comparator for patients with F508del/MF genotypes was placebo because there is no approved CFTR modulator available for these patients. In these phase 2 studies, VX-659/TEZ/IVA and VX-445/TEZ/IVA demonstrated clinically significant improvements in lung function as measured by the absolute change from baseline in forced expiratory volume in 1 s (FEV1) % pred through 4 weeks of treatment in patients with F508del/MF genotypes (within-group increases from baseline of up to 13.3 and 13.8 percentage points, respectively) and the F508del/F508del genotype (within-group increases from baseline of 9.7 and 11.0 percentage points beyond effects with TEZ/IVA, respectively) (figure 1) [25, 27]. In each case, as with other CFTR modulators, the vast majority of FEV1 % pred responses were present at 2 weeks [14–17], indicating that the improvements in lung function occurred rapidly. VX-659 and VX-445 triple combinations also reduced (i.e. improved) sweat chloride concentrations, an important in vivo pharmacodynamic biomarker of CFTR function, in patients with F508del/MF genotypes (within-group decreases from baseline up to −51.4 and −39.1 mmol·L−1, respectively) and the F508del/F508del genotype (within-group decreases from baseline of −42.2 and −39.6 mmol·L−1 beyond effects with TEZ/IVA, respectively) [25, 27]. Improvements in Cystic Fibrosis Questionnaire-Revised (CFQ-R) respiratory domain scores (for which the established minimal clinically important difference is 4.0 points) [36] were also robust with VX-659 and VX-445 triple combinations (within-group change from baseline up to 24.6 and 25.7 points in the F508del/MF cohorts; within-group change from baseline of 19.5 and 20.7 points beyond effects with TEZ/IVA in the F508del/F508del cohorts, respectively) and compared favourably with those observed in prior CFTR modulator studies. VX-659 and VX-445 triple combinations had acceptable safety profiles over 4 weeks, with the majority of adverse events being mild or moderate [25, 27]. These phase 2 proof-of-concept studies demonstrated that targeting the F508del-CFTR protein with a triple-combination regimen containing two complementary correctors and a potentiator can restore substantial CFTR function, even in patients with a single F508del-CFTR allele, leading to clinically significant improvements in lung function [25, 27]. Indeed, the magnitude of response with these triple-combination regimens exceeded the benchmark of robust improvement in CFTR modulation set by IVA in patients with a G551D-CFTR mutation [14, 25, 27].

Summary of absolute changes from baseline in a, c) forced expiratory volume in 1 s (FEV1) % pred and b, d) sweat chloride concentration after triple-combination treatment for 4 weeks with a, b) VX-659/tezacaftor (TEZ)/ivacaftor (IVA) or c, d) VX-445/TEZ/IVA in patients with F508del/minimal function (MF) or F508del/F508del genotypes evaluated in phase 2 studies [25, 27]. Patients with F508del/F508del received TEZ/IVA during a 4-week run-in period, thus absolute changes in this group were in addition to changes with TEZ/IVA alone. Dotted lines indicate absolute change from baseline in a, c) FEV1 % pred (10.4 percentage points) and b, d) sweat chloride concentration (−48.7 mmol·L−1) through week 24 with IVA in patients with a G551D mutation [14]. 95% confidence intervals are indicated.
These highly promising phase 2 results led to the advancement of VX-659 and VX-445 triple combinations to parallel phase 3 development programmes (ECLIPSE and AURORA, respectively). Together, the ECLIPSE and AURORA programmes represent one of the largest phase 3 CFTR modulator development programmes ever conducted in CF. The programmes were informed by previous experience with CFTR modulators, incorporating efficient study designs to rapidly identify a triple-combination regimen suitable as a new treatment option for patients with one or two F508del-CFTR mutations.
Each programme includes separate pivotal studies in patients with the F508del/MF and F508del/F508del genotypes. This approach is necessary because the standard of care is different in these two populations [25, 27] and, as mentioned earlier, they require different comparator groups (placebo plus TEZ/IVA for patients with the F508del/F508del genotype; placebo for patients with F508del/MF genotypes).
The studies in patients with F508del/MF genotypes (VX17-659-102 and VX17-445-102) serve as the placebo-controlled anchor studies for their respective triple-combination programmes. These are 24-week studies that will assess both short- and longer-term changes in lung function as well as longer-term changes in pulmonary exacerbations and nutritional status. The primary end-point is the absolute change in FEV1 % pred from baseline, assessed at week 4 (global protocol) and through week 24 (European protocol). These studies in patients with F508del/MF genotypes are also powered to assess the change in pulmonary exacerbation rates from baseline through week 24. Because studies in patients with the F508del/F508del genotype already receiving TEZ/IVA would require prohibitively large patient numbers (more than 1500 patients each) to be sufficiently powered to detect 30% reductions in pulmonary exacerbations over a 24-week treatment period, studies in patients with F508del/MF genotypes provide a unique opportunity to effectively assess pulmonary exacerbation rates in both the ECLIPSE and AURORA programmes. Importantly, experience with approved CFTR modulators across multiple studies has shown that the safety profile of CFTR modulators is independent of CFTR genotype; therefore, the studies in patients with F508del/MF genotypes will provide placebo-controlled safety data applicable to both F508del/MF and F508del/F508del populations.
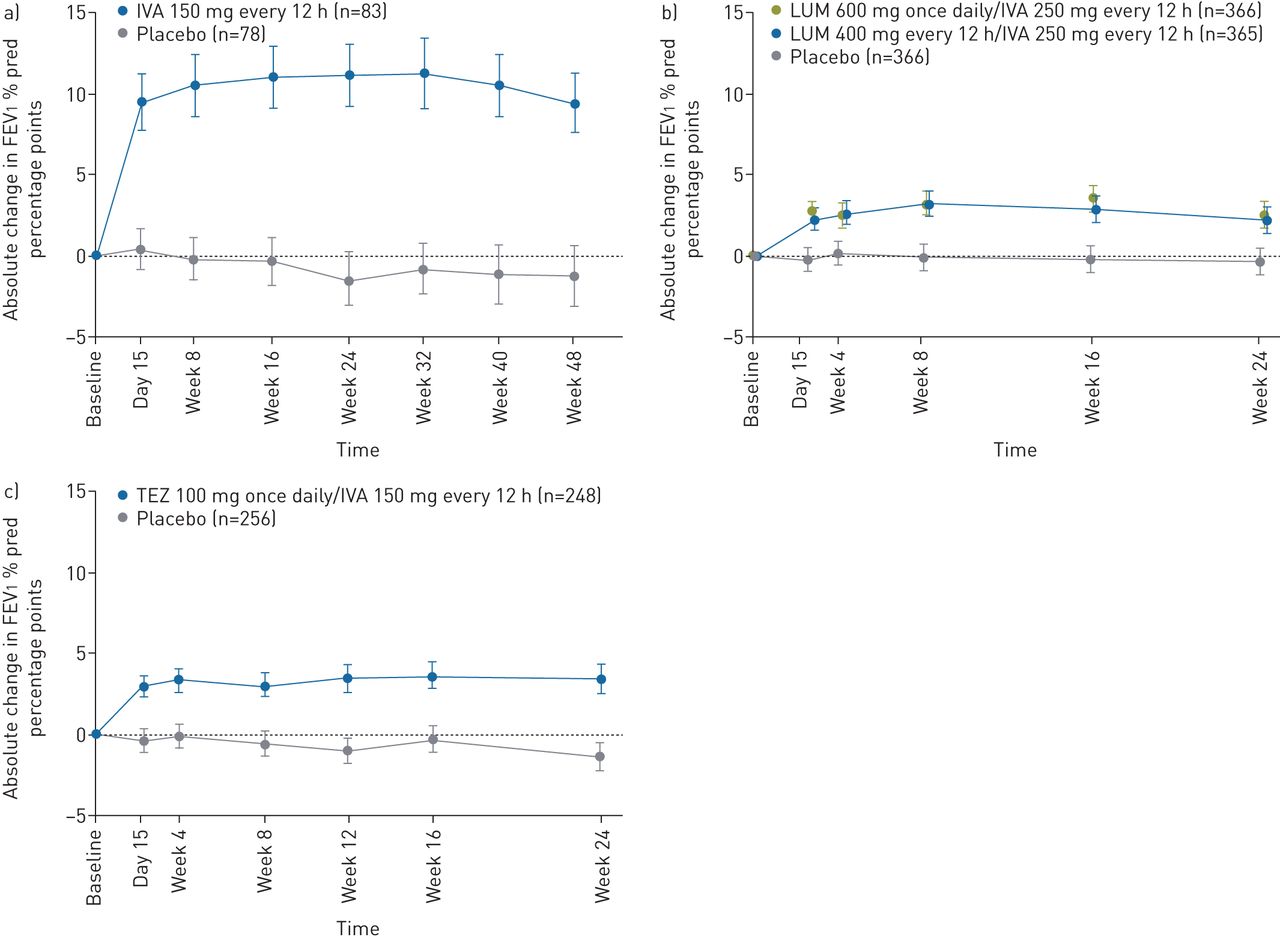
The studies in patients with the F508del/F508del genotype (VX17-659-103 and VX17-445-103) are 4 weeks in duration and powered to assess the primary end-point of absolute change in FEV1 % pred from baseline at week 4. The 4-week time-point was selected for evaluation of lung function across all studies because experience with the approved CFTR modulators IVA, LUM/IVA and TEZ/IVA has consistently shown that the absolute change in FEV1 % pred from baseline at 4 weeks predicts durable improvements in lung function, as illustrated by longitudinal plots showing the mean absolute change from baseline in FEV1 % pred through 24 to 48 weeks in representative phase 3 studies (figure 2). In all representative studies, there was rapid separation (i.e. by day 15) between the FEV1 % pred curves for the active treatment and placebo groups that was sustained [14–17, 37]. This rapid effect and the ability of short-term outcomes to predict long-term effects has been observed across groups of patients with different genotypes, baseline FEV1 % pred and age [14–17, 30, 37–39]. Importantly, lung function and other efficacy outcomes observed with approved CFTR modulators in pivotal trials have been sustained during long-term use [40, 41].
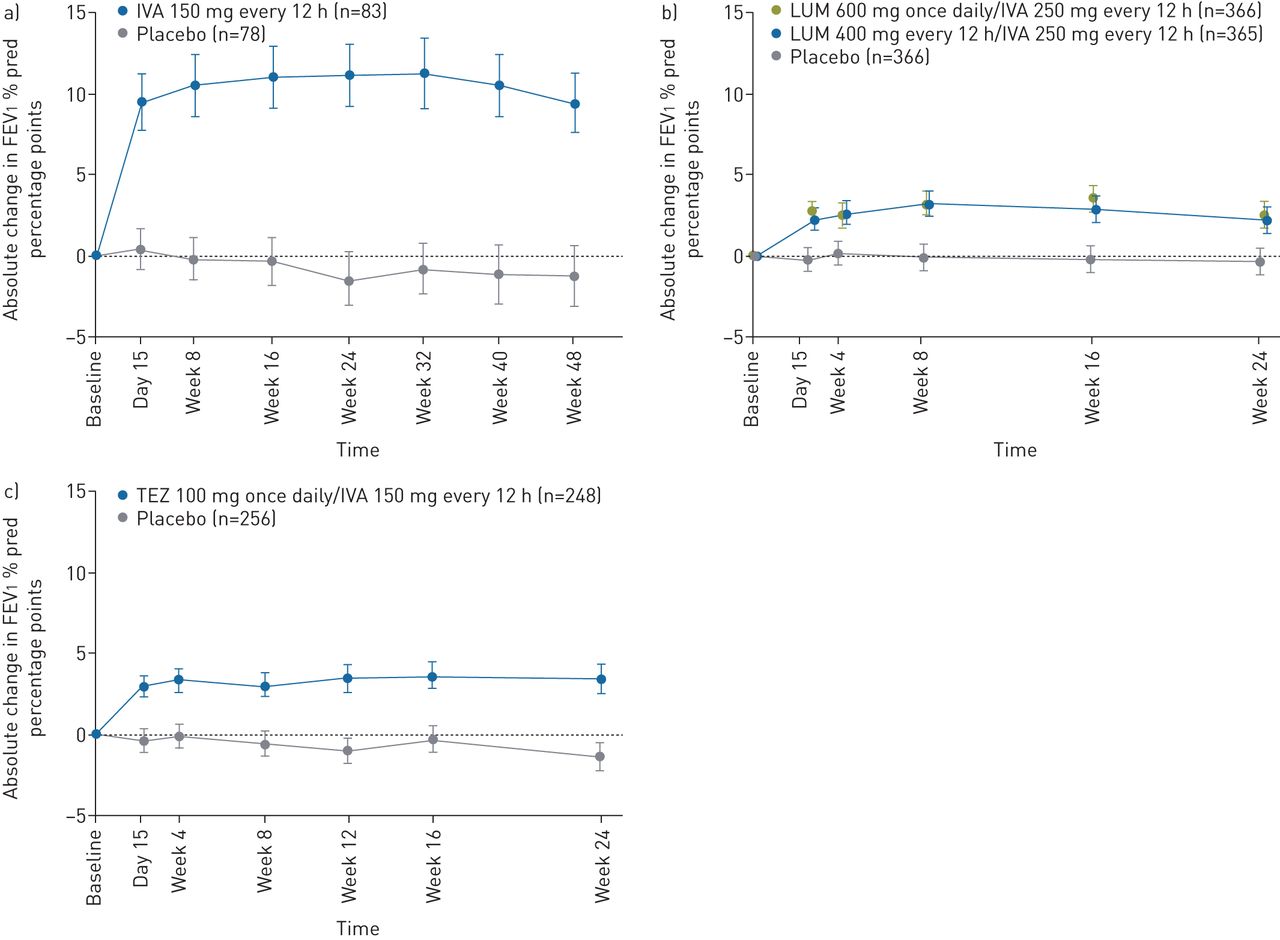
Absolute change in forced expiratory volume in 1 s (FEV1) % pred over time in clinical trials with a) ivacaftor (IVA) in patients with at least one G551D allele [14], b) lumacaftor (LUM)/IVA in patients with the F508del/F508del genotype [15] and c) tezacaftor (TEZ)/IVA in patients with the F508del/F508del genotype [16]. Reproduced from [14–16] with permission.
Long-term safety and efficacy will be assessed in the ECLIPSE and AURORA programmes through compound-specific 96-week open-label extension studies made available to patients following their completion of the randomised placebo-controlled (F508del/MF) or active-controlled (F508del/F508del) treatment studies.
Methods
Randomised controlled studies: VX17-659-102, VX17-445-102, VX17-659-103 and VX17-445-103
Study design and participants
VX17-659-102 and VX17-445-102 are two identically designed randomised, double-blind, placebo-controlled studies evaluating the efficacy and safety of VX-659 (VX17-659-102; ClinicalTrials.gov identifier NCT03447249) or VX-445 (VX17-445-102; ClinicalTrials.gov identifier NCT03525444) in triple combination with TEZ/IVA in patients with CF with F508del/MF genotypes (figure 3a) as previously defined [25, 27]. Mutations that are considered MF based on in vitro testing have baseline chloride transport that is <10% of wild-type CFTR and increases <10% over baseline after the addition of TEZ, IVA or TEZ/IVA [25, 42]. These studies were each planned to randomise approximately 360 patients (1:1) to triple combination (active treatment) or placebo (n=180 per group) for 24 weeks.

{kind=link}
{kind=link}
{kind=link}
Study design for ECLIPSE (VX-659 triple combination) and AURORA (VX-445 triple combination) phase 3 studies. TEZ: tezacaftor; IVA: ivacaftor; FEV1: forced expiratory volume in 1 s. a) Randomised controlled studies in patients with F508del/minimal function genotypes: VX17-659-102 (n∼360) and VX17-445-102 (n∼360). b) Randomised controlled studies in patients with the F508del/F508del genotype: VX17-659-103 (n∼100) and VX17-445-103 (n∼100). c) Open-label extension studies: VX17-659-105 (n∼400) and VX17-445-105 (n∼400). All sample sizes are per study. #: the 4-week safety follow-up visit in the parent studies is required only for patients who do not proceed to the open-label extension studies.
VX17-659-103 and VX17-445-103 are two identically designed randomised, double-blind studies evaluating the efficacy and safety of VX-659 (VX17-659-103; ClinicalTrials.gov identifier NCT03460990) or VX-445 (VX17-445-103; ClinicalTrials.gov identifier NCT03525548) in triple combination with TEZ/IVA compared with TEZ/IVA alone in patients with CF with the F508del/F508del genotype (figure 3b). These studies were each planned to randomise approximately 100 patients (1:1) to triple combination or TEZ/IVA (n=50 per group) for 4 weeks.
All studies include male and female patients aged ≥12 years with mild to moderate stable CF (FEV1 % pred ≥40% and ≤90% at screening). Additional inclusion and exclusion criteria are listed in table 1.
Key inclusion and exclusion criteria
All studies include a 4-week screening period, a treatment period and a 4-week safety follow-up period for patients not proceeding to an open-label study. The regimen administered during the treatment period includes triple combination with VX-659 (240 mg once daily) or VX-445 (200 mg once daily) plus the approved commercial dose of TEZ (100 mg once daily) and IVA (150 mg every 12 h), or matched placebo control (triple placebo in the F508del/MF cohorts or placebo plus TEZ/IVA in the F508del/F508del cohorts). Patients with the F508del/F508del genotype receive TEZ/IVA during a 4-week run-in period to maintain standard-of-care therapy and to provide a reliable baseline to assess the benefit of adding a second CFTR corrector to TEZ/IVA. All study drugs are administered orally. Scheduled clinical, laboratory and safety assessments are listed in tables 2 and 3.
Treatment periods and safety follow-up visits: randomised controlled studies in patients with F508del/MF genotypes: VX17-659-102 and VX17-445-102
Treatment periods and safety follow-up visits: randomised controlled studies in patients with the F508del/F508del genotype: VX17-659-103 and VX17-445-103
Randomisation and masking
Patients are randomised 1:1 after screening (F508del/MF genotypes) or after the TEZ/IVA run-in (F508del/F508del genotype). Patients are stratified by FEV1 % pred (<70% versus ≥70%) and age (<18 versus ≥18 years); patients with F508del/MF genotypes are also stratified by sex (male versus female). Treatment is assigned with an interactive web-based response system and is blinded for all patients, caregivers, investigators, site personnel and the sponsor (i.e. the study team).
Study end-points
The primary objective of each study is to evaluate the efficacy of VX-659 or VX-445 in triple combination with TEZ/IVA in patients with CF with F508del/MF or F508del/F508del genotypes. In the global protocols, the primary end-point in patients with F508del/MF and F508del/F508del genotypes is absolute change in FEV1 % pred from baseline at week 4. In the European protocol, the primary end-point in patients with F508del/MF genotypes is absolute change in FEV1 % pred from baseline through week 24. Key secondary and other end-points are listed in table 4.
Study end-points
Statistical analyses
The absolute change from baseline in FEV1 % pred at week 4 will be evaluated in all studies, with the change through week 24 also evaluated in studies in patients with F508del/MF genotypes. These end-points will be analysed using a mixed effects model for repeated measures (MMRM) with change from baseline in FEV1 % pred as the dependent variable. The model will include treatment group, visit and treatment-by-visit interaction as fixed effects, with continuous baseline FEV1 % pred and age (<18 versus ≥18 years) as covariates; sex (female versus male) will be included as a covariate in studies in patients with F508del/MF genotypes, and the model will use an unstructured covariance for the within-subject errors. The MMRM model has been used successfully throughout much of the CFTR modulator development programme and provides advantages such as evaluating a treatment effect throughout the study period, handling for missing data and reducing bias [43].
In the 24-week studies in patients with F508del/MF genotypes, an interim analysis of efficacy (including absolute change from baseline in FEV1 % pred) and safety is planned after 140 or more patients complete the week 4 visit and 100 or more patients complete the week 12 visit, permitting the option for accelerated regulatory submissions in selected regions. A Lan–DeMets α spending function will be applied to control the overall type I error rate of 0.05 for the 4-week end-point during the interim analysis and the final analysis, such that an α of 0.01 will be preserved for the final analysis [44]. If the number of patients included in the interim analysis is 140, then the absolute change from baseline in FEV1 % pred at week 4 will be tested at a significance level of 0.044 during the interim analysis. The actual α at the interim analysis (α0) will be determined based on the actual number of patients included in the interim analysis. Assuming a within-group standard deviation of 7 percentage points and a 5% dropout rate at week 4, an interim analysis sample size of 70 patients in each treatment group will have ∼98% power to detect a difference of 5 percentage points between the treatment groups, based on a two-sided, two-sample t-test at a significance level of 0.044. Regardless of the outcome of the interim analysis, the study will remain blinded, and all patients will continue through the 24-week treatment period.
Approximately 360 patients with F508del/MF genotypes per study were determined to provide adequate power to detect a difference in the number of pulmonary exacerbation events between triple combination and placebo through week 24. Assuming a pulmonary exacerbation rate of 0.6 for the triple-placebo groups over 24 weeks and accounting for a 10% dropout rate, based on a two-sided, two-sample negative binomial regression model test for the ratio of rates at a significance level of 0.05, the power to detect a 40% reduction in pulmonary exacerbation rate for triple-combination groups compared with placebo groups in these studies is ∼80%. To adjust for multiplicity, pulmonary exacerbations and other key secondary end-points will be tested only at the final analysis and only if the primary end-point shows statistical significance.
In studies in patients with the F508del/F508del genotype, assuming a within-group standard deviation of 7 percentage points and accounting for a 5% dropout rate at week 4, based on a two-sided, two-sample t-test at a significance level of 0.05, a sample size of 50 patients per treatment group is expected to have ∼93% power to detect a difference of 5 percentage points in the mean absolute change in FEV1 % pred from baseline at week 4.
In these studies, some additional end-points, such as absolute change in CFQ-R respiratory domain score, will be analysed using an MMRM model similar to that used for the primary end-point. The full analysis set will be used to assess efficacy and will include all randomised patients with the intended CFTR genotype who received one or more doses of study drug. The safety analysis will include all patients who received one or more doses of study drug.
Open-label extension studies: VX17-659-105 and VX17-445-105
Provided patients have not permanently discontinued the study drug in the parent study, patients who complete the randomised treatment periods for VX-659 or VX-445 in triple combination with TEZ/IVA (VX17-659-102 or VX17-659-103 and VX17-445-102 or VX17-445-103, respectively) will have the option to enrol in the respective open-label extension study VX17-659-105 (ClinicalTrials.gov identifier NCT03447262) or VX17-445-105 (ClinicalTrials.gov identifier NCT03525574) (table 1 and figure 3a and b). Each open-label extension study will include over 400 patients with eligible genotypes (F508del/MF or F508del/F508del). The primary end-point of the open-label extension studies is the safety and tolerability of long-term treatment with VX-659 or VX-445 in triple combination with TEZ/IVA. Evaluation of long-term efficacy is a secondary objective (table 4).
Each open-label extension study consists of a 96-week treatment period, during which patients receive the same dose of VX-659 or VX-445 triple combination used in the respective parent study, followed by a safety follow-up visit 4 weeks after the last dose of study drug. Safety will be evaluated in all patients who received one or more doses of study drug (table 5 and figure 3c).
Treatment periods and safety follow-up visits: open-label extension studies: VX17-659-105 and VX17-445-105
Ethical approval and patient consent
All studies will be conducted in accordance with the International Council for Harmonisation Good Clinical Practice guidelines, consistent with the principles of the Declaration of Helsinki. Study documentation will be approved by institutional review boards/independent ethics committees before study initiation. Written informed consent (and assent, if applicable) will be obtained before participation.
Discussion
Here we describe remarkable progress from the pre-clinical stage to global phase 3 programmes evaluating two next-generation CFTR correctors, VX-659 and VX-445, in triple combination with the first-generation corrector TEZ and the potentiator IVA. Many factors contributed to the ability to design and conduct these unique programmes. First, understanding the multiple molecular deficits that result from the F508del-CFTR mutation led to the hypothesis that the response of the mutant protein to existing corrector–potentiator regimens could be further enhanced with the addition of a second corrector [25, 27]. Second, the strategy was based on a strong pre-clinical model that has been highly predictive of clinical results [9, 14]. Previous experience with in vitro HBE results translating into clinical improvement in FEV1 % pred and other clinical outcomes provided confidence that triple combinations would be efficacious in patients with an F508del/MF or F508del/F508del genotype, a confidence that was reinforced by phase 2 results consistent with predicted estimates of clinical outcome [25, 27]. Third, prior experience with CFTR modulator clinical development programmes provided the basis for an efficient development strategy, including a 4-week primary efficacy end-point, that will potentially enable an expedited drug development programme in selected regions.
Cumulative experience with effective CFTR modulators allowed for the design of complementary studies in each of the triple-combination programmes, leveraging the 24-week placebo-controlled design in patients with F508del/MF genotypes to obtain safety data applicable to the F508del/MF and F508del/F508del populations and to provide long-term placebo-controlled data on the number of pulmonary exacerbation events and other outcomes. Finally, these phase 3 programmes could not have been successfully conducted without the support of the Cystic Fibrosis Therapeutics Development Network in the USA and the European Cystic Fibrosis Clinical Trials Network, agreement and input from investigative teams, and willingness of patients to participate in the trials. Outreach, co-development and communication among the stakeholders enabled efficient trial design and conduct, while safety, consistency, and trial and data integrity were maintained. Together, these studies represent the culmination of a decade of pre-clinical and clinical experience with CFTR modulators to address the underlying protein defect caused by the F508del-CFTR mutation and to determine whether a triple-combination regimen can provide meaningful clinical benefits to the majority of patients with CF.
Conclusions
Extensive CFTR modulator experience was leveraged to design and conduct an unprecedented, efficient and data-driven phase 3 development programme investigating two triple-combination regimens in parallel in patients with one or two F508del-CFTR alleles. Data generated from this programme have the potential to demonstrate the substantial advancement in therapy that triple combinations provide over current therapies in both patient populations, by providing an effective CFTR modulator therapy for patients for whom no CFTR modulators are currently approved (i.e. F508del/MF genotypes) and an enhanced CFTR modulator therapy for patients with the F508del/F508del genotype. Approximately 90% of people with CF carry one or more copies of the F508del-CFTR allele, and this programme represents an important step towards potentially treating the underlying protein defect in these patients with a single highly effective triple-combination regimen.
Acknowledgements
The authors wish to thank the patients and their families, the trial coordinators, and the investigators for their contributions to the trials. Sarah Garber (Vertex Pharmaceuticals Inc., Boston, MA, USA) provided editorial coordination and assistance. Katherine Mills-Luján (ArticulateScience LLC, Atlanta, GA, USA) provided editorial assistance under the direction of the authors, with support from Vertex Pharmaceuticals Inc.
Footnotes
This article does not report study data, but provides the rationale and study design for two comprehensive phase 3 development programmes (ECLIPSE and AURORA): ClinicalTrials.gov identifiers NCT03447249, NCT03460990, NCT03447262, NCT03525444, NCT03525548 and NCT03525574.
Conflict of interest: J.L. Taylor-Cousar reports grants and personal fees from Vertex, during the conduct of the study; grants and personal fees from Vertex, Gilead, Celtaxsys and Proteostasis, grants from N30 and Bayer, personal fees from Novartis, Genentech, Protalix and Santhera, outside the submitted work.
Conflict of interest: M.A. Mall reports personal fees and nonfinancial support from Vertex, during the conduct of the study; grants from the German Federal Ministry of Education and Research (BMBF) and the Einstein Foundation Berlin, and personal fees from Arrowhead Pharmaceuticals, Bayer, Boehringer Ingelheim, Polyphor, ProQR Therapeutics, PTC Therapeutics, Spyryx Biosciences and Vertex, outside the submitted work.
Conflict of interest: B.W. Ramsey reports grants from Vertex, during the conduct of the study.
Conflict of interest: E.F. McKone reports grants and personal fees from Vertex, during the conduct of the study; personal fees from Proteostasis, grants from Gilead, and nonfinancial support from Novartis, outside the submitted work.
Conflict of interest: E. Tullis reports grants, personal fees and nonfinancial support from Vertex, during the conduct of the study; grants, personal fees and nonfinancial support from Proteostasis and Spyryx, and grants from Corbus and Celtaxys, outside the submitted work.
Conflict of interest: G. Marigowda is an employee of Vertex and may hold stock and/or stock options in Vertex; he reports other funding from Vertex during the conduct of the study.
Conflict of interest: C.M. McKee is an employee and stockholder of Vertex; she reports other funding from Vertex during the conduct of the study.
Conflict of interest: D. Waltz is an employee of Vertex and may hold stock and/or stock options in Vertex; he reports other funding from Vertex during the conduct of the study. He is listed as an inventor on three pending patents: Methods of Treatment of Cystic Fibrosis, Methods of Treatment of Cystic Fibrosis, and Pharmaceutical Compositions for Treating Cystic Fibrosis.
Conflict of interest: S.M. Moskowitz is an employee of Vertex and may hold stock and/or stock options in Vertex. He has three patents pending: Methods of Treatment for Cystic Fibrosis, Methods of Treatment of Cystic Fibrosis and Pharmaceutical Compositions for Treating Cystic Fibrosis.
Conflict of interest: J. Savage is an employee of Vertex and may hold stock and/or stock options in Vertex; she reports other funding from Vertex during the conduct of the study.
Conflict of interest: F. Xuan is an employee of Vertex and may hold stock and/or stock options in Vertex; she reports other funding from Vertex during the conduct of the study.
Conflict of interest: S.M. Rowe reports grants and personal fees from Vertex, during the conduct of the study; and grants and personal fees from Bayer, Novartis and Celtaxsys; grants from Forest Research Institute, AstraZeneca, N30/Nivalis, Galapagos/AbbVie, Proteostasis, Eloxx and PTC; and personal fees from Renovion, outside the submitted work.
Support statement: This study was supported by Vertex Pharmaceuticals Inc., the National Institute of Diabetes and Digestive and Kidney Diseases (P30DK072482), and the National Heart, Lung, and Blood Institute (R35HL135816). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 8, 2019.
- Accepted April 19, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









