





Abstract
Idiopathic pulmonary fibrosis (IPF), the scarring of lung parenchyma resulting in the loss of lung function, remains a fatal disease with a significant unmet medical need. Patients with severe IPF often develop acute exacerbations resulting in the rapid deterioration of lung function, requiring transplantation. Understanding the pathophysiological mechanisms contributing to IPF is key to develop novel therapeutic approaches for end-stage disease.
We report here RNA-sequencing analyses of lung tissues from a cohort of patients with transplant-stage IPF (n=36), compared with acute lung injury (ALI) (n=11) and nondisease controls (n=19), that reveal a robust gene expression signature unique to end-stage IPF. In addition to extracellular matrix remodelling pathways, we identified pathways associated with T-cell infiltration/activation, tumour development, and cholesterol homeostasis, as well as novel alternatively spliced transcripts that are differentially regulated in the advanced IPF lung versus ALI or nondisease controls. Additionally, we show a subset of genes that are correlated with percent predicted forced vital capacity and could reflect disease severity.
Our results establish a robust transcriptomic fingerprint of an advanced IPF lung that is distinct from previously reported microarray signatures of moderate, stable or progressive IPF and identifies hitherto unknown candidate targets and pathways for therapeutic intervention in late-stage IPF as well as biomarkers to characterise disease progression and enable patient stratification.
Abstract
An RNA-Seq-based transcriptomic fingerprint of severe IPF enriched in pathways of T-cell infiltration/activation, tumour development and cholesterol homeostasis highlights novel splice variants, candidate targets and biomarkers in advanced IPF http://bit.ly/2YbTOv8
Introduction
Idiopathic pulmonary fibrosis (IPF) is a fatal disease of unknown aetiology characterised by the scarring of the lung parenchyma, resulting in the progressive loss of lung function and eventual death [1]. Although two recently approved medications for IPF (pirfenidone (Esbriet) and nintedanib (Ofev)) modestly reduce lung function decline in moderate IPF, they do not halt or reverse fibrosis, and do not significantly improve quality of life [2–4]. Lung transplant still remains the only option to prolong survival in patients with severe IPF [5]. Therapeutic approaches to IPF targeting numerous inflammatory and tissue remodelling pathways have consistently failed in the clinic, in part due to limited disease understanding, and lack of predictive diagnostic/prognostic biomarkers. Several studies in the past have utilised microarray profiling [6–9] and more recently single-cell RNA sequencing [10, 11] of IPF patient-derived lung tissue to identify genes and/or pathways differentially regulated in comparison with controls or patients with other lung diseases, providing signatures for disease classification. Peripheral blood profiling across small cohorts of patients with IPF has also identified potential biomarkers of disease [12–15]. Where available, gene/protein expression profiles have been associated with clinical diagnosis, disease severity and measures of lung function [8]. While these studies have shed light on pathways that could contribute to early, stable or progressive IPF, our knowledge of the pathways and mechanisms that contribute to severe/end-stage IPF remains very limited. Importantly, therapies targeting pathways identified to be dysregulated in patients with early/stable/progressive IPF have not been effective in the treatment of advanced IPF. Patients with severe IPF often develop additional lung complications including acute exacerbations, lung cancer and rapid decline in lung function, requiring lung transplantation [16]. The diagnosis of IPF often occurs very late in the clinical course, when the disease has progressed significantly. Thus, understanding of molecular mechanisms in severe IPF could help develop targeted therapies and personalised medicine approaches for this deadly disease.
We hypothesised that the molecular signature of severe IPF would be different from that of ALI and healthy controls and therefore, could help differentiate and stratify patients with advanced disease and identify novel therapeutic targets and biomarkers. Accordingly, we performed RNA sequencing on lung tissues from a cohort of patients with severe IPF that underwent lung transplantation (n=36) and compared this with tissues from nondiseased controls (n=19) and patients with clinical and pathological acute lung injury (ALI) (n=11). Furthermore, we used regression analyses to identify genes most strongly associated with lung function. Additionally, we identified alternative splicing of a large number of genes in advanced IPF. Using these complementary approaches, we established a robust transcriptomic fingerprint of severe IPF, revealed several key pathways (T-cell infiltration, immune response, host defence, cholesterol homeostasis and prostaglandin synthesis) that are differentially regulated and highlighted candidate biomarkers and targets as well as alternative isoform regulation. Our work therefore is important in expanding the knowledge of uniquely altered pathways that could lead to lung function decline in end-stage IPF and the identification of new biomarkers to predict organ failure and potential targets to treat advanced IPF.
Materials and methods
Human subjects and lung tissue acquisition
All human subject sample acquisitions (described in supplementary methods) and experiments were conducted with the appropriate approval from the Institutional Review Board (IRB 806468, IRB 813685). The clinical profile and demographics of IPF, ALI and control subjects are listed in table 1. Details of the acquisition protocol are provided in the supplementary material.
Profiles of control donor, idiopathic pulmonary fibrosis (IPF) and acute lung injury (ALI) patient cohorts used in the study
RNA sequencing
Illumina TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero (cat. No RS-122-2203, Illumina Inc., San Diego, CA) was used to generate sequencing libraries per manufacturer's recommendation. Gene expression was determined via RNA-Seq libraries run on an Illumina HiSeq 25 000 platform producing 75 bp paired-end (PE) reads. We generated on average 40 million PE reads for each sample. Reads were aligned to the human genome (GRch38) with the Omicsoft Sequence Aligner [17]. Gene and transcript abundance was determined using Ensembl release 90 human gene models [18] using RSEM [19]. All the gene expression data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GSE 134692).
RNA-Seq data analyses
All RNA-Seq data were processed in R with the Bioconductor packages [20]. RNA-Seq samples were TMM (Trimmed Mean of M Values) normalised [21] with the edgeR package [22]. Outlier detection was performed using t-distributed stochastic neighbour embedding (t-SNE) [23]. Contributions of known variables (disease state, age, sex) to the data variance were assessed by principal variance component analyses. Differential gene expression contrasts between treatment groups was performed using the limma package [24]. Pathway enrichment was computed using Genego (Metacore) and MetaBase (Clarivate Analytics) version 6.34.69200. We also performed ensemble gene set enrichment analyses using the molecular signature database (MSigDB), a comprehensive database from the Broad Institute encompassing numerous curated public gene sets and pathways [25].
Alternative isoform regulation
We used JunctionSeq software for detecting the differential usage of exons and splice junctions from RNA-Seq data [26]. This enabled the determination of alternative isoform regulation. Testable loci identified through JunctionSeq were further filtered by excluding those with mean counts ≥10 and an adjusted p-value ≤0.05.
Statistical analyses
Total gene expression data were analysed using a fold change/false discovery rate (FDR) cut-off of 2X/0.001 (for comparing IPF and control) or 1.5X/0.1 (for comparing IPF versus control and ALI versus control signatures) to generate gene lists for pathway analyses. Individual gene expression was compared across groups by one-way ANOVA followed by Tukey's post-test with differences considered statistically significant at p≤0.05.
Results
t-SNE variance analyses
We used the t-SNE method to visualise the distribution of the expression data across control and diseased samples and identify outliers. As shown in figure 1a, there was a clear separation of the IPF samples from both the control and ALI samples. To assess the contribution of donor variables to gene expression, we performed variance partition analyses. Figure 1b shows that the largest contributor to the variance among known variables was the IPF or control disease state reaffirming that the difference in normalised gene expression was driven primarily by the IPF or control disease state (“Residual” represents a residual sum of unknown variables and could include polypharmacology, medications, and genetic factors among many others). Other known parameters such as age (although significantly higher in IPF), sex or ethnicity did not appear to significantly contribute towards the changes in expression.

Quality control analyses of RNA-sequencing data from idiopathic pulmonary fibrosis (IPF) (n=36), acute lung injury (ALI) (n=10) and control (n=20) lung tissue. a) Principal component analyses of batch-corrected normalised expression data using the t-scholastic neighbourhood enrichment (t-SNE method). b) Principal variance component analyses were used to assess the relative contribution of patient variables to variance in the normalised expression across the samples.
Differential gene expression and pathway regulation in IPF
Figure 2a shows the heatmap of scaled gene expression of the samples generated by hierarchical clustering and represents a transcriptomic fingerprint of advanced IPF. Greater than 90% of the IPF samples clearly clustered together confirming that they represent a unique cohort of patients with a distinct molecular signature. Given the robust separation of the IPF samples from both ALI and control samples, we first analysed the IPF fingerprint through Metacore pathway analyses using a stringent cut-off of fold change ≥2 and an adjusted p-value ≤0.001. Consistent with an advanced disease state, we identified a strong upregulation of pathways associated with tumour cell infiltration and development of cancer (figure 3a) in addition to expected alterations in extracellular matrix (ECM) remodelling pathways. Notably, T-cell activation and survival pathways (CD4, CD8 and regulatory T-cells) were strongly upregulated concomitant with a robust increase in the expression of the checkpoint effectors programmed cell death (PD)1, LAG3 and CTLA4, the T-cell costimulatory receptor CD28 as well as chemokine/chemokine receptors including CCR5, CCR6, CXCR3 and CXCR5 (figure 3b). The T-cell signature in this cohort was more prominent than the classical or alternatively activated monocyte/macrophage signature. Of note, genes involved in inflammatory signalling leading to myeloid-derived suppressor cells or M2 macrophages were strongly downregulated (figure 3c; calgranulin/S100A8/S100A9, RAGE and TLR4, although TLR4 did not meet the stringent cut-off for this analysis).

Transcriptomic fingerprint of advanced idiopathic pulmonary fibrosis (IPF) lung. Differential gene expression contrasts between treatment groups was performed using the limma package. Heatmap showing scaled intensity of individual genes in rows and samples in columns. Note samples predominantly cluster based on disease status. ALI: acute lung injury; F: female; M: male; U: unknown.
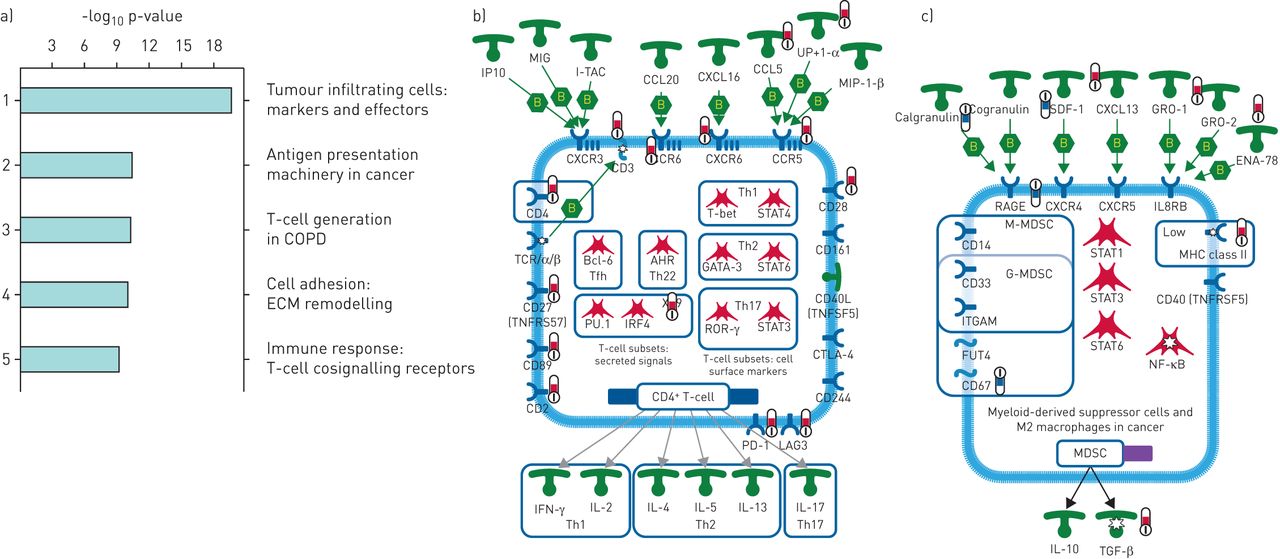
Differential pathway regulation in advanced idiopathic pulmonary fibrosis (IPF) lung. a) Metacore analyses of pathway enrichment in IPF versus control lungs using a filter of positive and negative fold change ≥2 and adjusted p-value of ≤0.001. b) Genego pathway map of tumour-infiltrating T-cells showing selected markers and key effector molecules. c) Genego pathway map of myeloid-derived suppressor cells in cancer. Red and blue thermometers indicate pathway components showing increased or decreased expression in IPF respectively. ECM: extracellular matrix; IP10: 10-kDa interferon-γ-inducible protein; MIG: monokine induced by interferon-γ; I-TAC: interferon-inducible T-cell α-chemoattractant; MIP: macrophage inflammatory protein; Th: T-helper; STAT: signal transducer and activator of transcription; TCR: T-cell receptor; TNFR: tumour necrosis factor receptor; Tfh: T-follicular helper; IRF: interferon regulatory factor; ROR: RAR-related orphan receptor; CTLA: cytotoxic T-lymphocyte-associated protein; PD: programmed death receptor; LAG: lymphocyte activation gene product; IFN: interferon; IL: interleukin; SDF: stromal cell-derived factor; GRO: growth-related oncogene protein; ENA: epithelium-derived neutrophil-activating protein; RAGE: receptor of advanced glycosylation end-products; IL8R: interleukin-8 receptor; M-MDSC: monocytic myeloid-derived suppressor cell; G-MDSC: granulocytic myeloid-derived suppressor cell; ITGAM: integrin subunit αM; FUT: fucosyltransferase; MHC: major histocompatibility complex; TNFRSF: tumour necrosis factor receptor superfamily; NF-κB: nuclear factor, κ-light chain enhancer of activated B-cells; TGF: transforming growth factor.
Comparison of pathway regulation in IPF and ALI patient cohorts
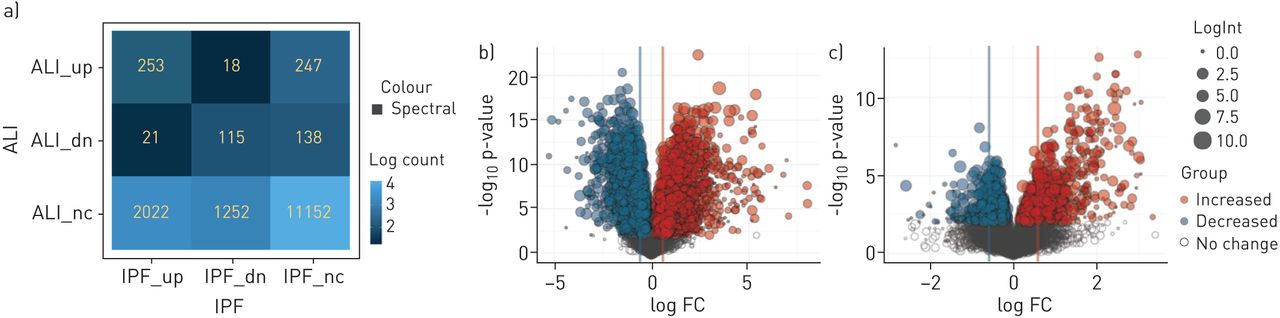
We employed multiple methods to identify the key genes and pathways that were uniquely regulated in the IPF and ALI patient cohorts. The complete list of gene changes for the different contrasts is provided in Supplementary Table T1. Figure 4 shows a contingency matrix of the number of genes that were significantly modulated across the different comparison groups with a fold change/adjusted p-value cut-off of 1.5/0.1. The IPF group compared with non-diseased controls expectedly showed the maximum number of gene changes, and genes commonly regulated in ALI and IPF mostly changed in the same direction. The volcano plots (figure 4b) further confirm that the number and magnitude of gene changes were far higher in the IPF versus control contrast. Genes uniquely regulated in each of these cohorts were analysed using the MetaBase pathway analyses tool. Immune response and T-cell activation pathways were strongly upregulated in IPF when compared with ALI, in addition to several expected fibrotic pathways including cell adhesion, ECM remodelling and epithelial mesenchymal transition (EMT) (figure 5a). Surprisingly, we also found that the cholesterol homeostasis pathway was strongly and significantly downregulated in IPF compared with both controls. In contrast, analyses of gene subsets unique to ALI revealed a strong regulation of cell cycle-associated pathways (Figure 5b). Importantly, within commonly regulated pathways across cohorts, we noted that a relatively greater fraction of genes within the “T-cell cosignalling pathway” and “Cell adhesion-remodelling” pathway were altered in IPF suggesting increased involvement of these genes/pathways in advanced IPF. Further analyses of the data with the Msigdb tool confirmed many of these findings (supplementary figure S1) and additionally identified a subset of robustly downregulated genes within the cholesterol homeostasis pathway overlapping with statin-regulated genes in the human lung (supplementary figure S2).

Comparison of gene expression changes in idiopathic pulmonary fibrosis (IPF) versus control and acute lung injury (ALI) versus control contrasts. Differential gene expression contrasts between treatment groups was performed using the limma package. Gene lists were generated using a cut-off of positive and negative fold change ≥1.5 and adjusted p-value of ≤0.1. a) Contingency table shows numbers and direction of movement of differentially altered genes and a measure of overlap/divergence between contrast groups. b) and c) Volcano plots showing the number and magnitude of significant gene changes across the IPF versus control and ALI versus control contrasts, respectively.
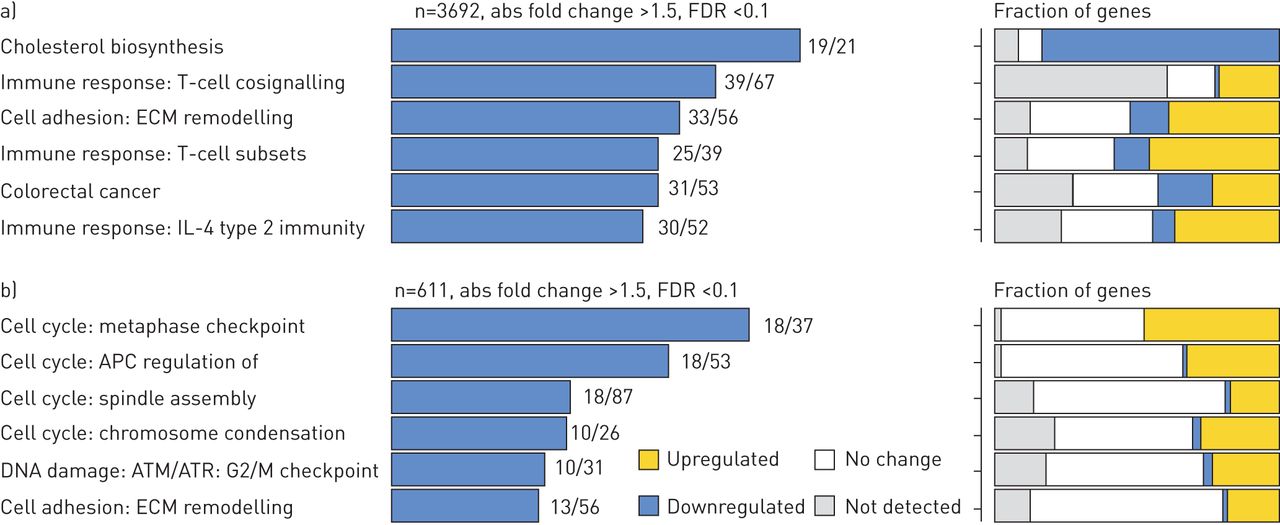
Comparison of pathway enrichment in a) idiopathic pulmonary fibrosis (IPF) and b) acute lung injury (ALI) cohorts. Gene lists were generated using a cut-off of positive and negative fold change ≥1.5 and adjusted p-value of ≤0.1. Left panels: pathways uniquely enriched in the IPF or ALI cohort. Numbers against each bar represent number of differentially altered genes compared with the total number of genes defining that pathway. Right panels: fraction of genes within the pathway that show increase, decrease or no alteration. ECM: extracellular matrix; FDR: false discovery rate.
Since the patients in the IPF cohort in this study were significantly older compared with controls or the ALI cohort, we performed the analyses with the inclusion of age in the statistical model (see supplementary material figures S3 and S4); however, such inclusion did not significantly change the findings with respect to variance or pathway analyses.
Alternative isoform regulation in IPF tissue
An important advantage of using RNA-Seq is the ability to detect gene splicing, which is not as accessible with microarray platforms. Accordingly, we analysed the gene expression data using the “JunctionSeq” tool to identify splice variants and differentially regulated exons to provide a measure of alternative isoform regulation. After applying count threshold and FDR filters, we identified 2723 exon junctions that were differentially regulated in IPF lung tissue. The top 15 loci showing the strongest isoform regulation are shown in figure 6a (the full list of differentially regulated loci is available in supplementary table T2). A Metacore analysis of genes with significantly altered exon junctions revealed several fibrosis-relevant pathways including cytoskeleton remodelling, cell adhesion, transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF) signalling. Interestingly, pathways involved in lysophosphatidic acid (LPA)-mediated G protein-coupled receptor signalling were also enriched (figure 6b). In comparison with the only RNA-Seq data published previously with a small cohort of IPF lung tissue (n=8, Nance et al., 2014 [27]), we identified a three-fold greater number of significantly regulated loci with 83 genes identified in common across these two studies (figure 7a). Importantly, several of the common genes are implicated in or associated with fibrosis. Wnt2b, a common gene in both our study and that of Nance et al., appeared to show a significantly altered expression of multiple exons, while the overall expression of the gene was unchanged (figure 7b). Moreover, the altered exonic expression appears to map to a specific transcript, suggesting this transcript is expressed differentially in patients with IPF.

Alternative isoform regulation in advanced idiopathic pulmonary fibrosis (IPF) lung. a) List of top loci that show alternative splicing in the IPF lung analysed with the ‘JunctionSeq’ tool. b) Metacore analyses of pathways enriched by genes that show significant alternative isoform/exon usage in the IPF lung (adjusted p-value ≤0.05). GPCR: G-protein coupled receptor; IL: interleukin; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; TGF: transforming growth factor.

a) Comparison of alternative transcripts identified in idiopathic pulmonary fibrosis (IPF) lung in our study (Upenn IPF) versus those identified in a published study by Nance et al. (2014). b) Alternative isoform usage in Wnt2b gene. Note the higher expression of certain exons and lower expression of others in the IPF lung.
Association between gene expression and lung function
We correlated gene expression to % predicted forced vital capacity (FVC), as a marker of disease severity, using Spearman's correlation analyses for nonlinear association studies. We identified nearly 300 genes that were significantly correlated with % predicted FVC (supplementary table T3). The top 15 genes that correlated positively with a decline in lung function are shown in figure 8a, and a Metacore pathway analyses that indicated that the gene subsets were enriched in fibrosis-relevant pathways are shown in figure 8b. The correlation of 2 of the top 15 genes (secretogranin-2 and semaphorin-3C) presented in figure 8c–e demonstrates that their expression in IPF tissue was also significantly elevated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation of idiopathic pulmonary fibrosis (IPF) lung gene expression to lung function. a) List of top genes that show significant inverse correlation to % predicted forced vital capacity (FVC) as assessed by Spearmen's correlation analyses. b) Metacore analyses of pathways enriched by genes that show significant correlation with % predicted FVC (adjusted p-value ≤0.05). c) and d) Correlation plots of gene expression of SCG2 and SEMA3C, two candidate genes identified through Spearman's analyses as significantly correlated with % predicted FVC. e) Gene expression intensity of SCG2 and SEMA3C confirming significant increase of these genes in IPF lung compared with control lung. tRNA: transfer RNA; GPCR: G-protein coupled receptor; TGF: transforming growth factor; ECM: extracellular matrix; BMP: bone morphogenetic protein; FVC: forced vital capacity.
Discussion
We present here a markedly distinct transcriptomic fingerprint of advanced IPF obtained through RNA-sequencing analysis of a cohort of patients with transplant-stage IPF in comparison with non-diseased controls and patients with ALI. In addition to previously reported pathways involved in epithelial injury and ECM homeostasis, our study identified several novel pathways and candidate genes that are altered in end-stage disease, including T-cell infiltration/activation pathways, cell–matrix interactions, cholesterol homeostasis and steroid biosynthesis. We also demonstrate extensive alternative transcript/exon/isoform usage in IPF, identifying novel splice variants that may be involved in disease pathology. Additionally, we show a novel subset of genes whose expression appears to be correlated with lung function decline in advanced IPF.
The molecular signature of our IPF cohort is distinctly different from past studies that have used lung tissues from patients with IPF with mild-to-moderate disease. Yang et al. [9] investigated the transcriptomic profile of IPF/usual interstitial pneumonia from the Lung Tissue Research Consortium and identified subgroups of patients with alterations in cilium-associated genes and epithelial injury pathways, further validating their findings with samples from the National Jewish Health cohort. Notably, the mean % FVC in their test cohort was ≥59%. In contrast, the mean % FVC of patients in our study was 44%, with a majority of patients at ≤30%. Likewise, microarray studies by Depianto et al., with a cohort of 40 patients with IPF, identified two primary clusters of coregulated genes representing bronchiolar epithelium and lymphoid aggregates [28]. Worsening IPF pathology remains a significant risk factor for acute exacerbations, development of lung cancer and additional lung complications that warrant a lung transplant. Konishi et al. [6] showed that the gene expression profile of acute exacerbations of IPF (AE-IPF) was mostly similar to that of stable IPF with only a few genes significantly altered between the two groups. Interestingly, these authors also observed a downregulation of AGER (receptor for advanced glycation end products) as we noted in our cohort of advanced IPF. However, defensin a3 (DEFA3) was strongly reduced in our cohort in contrast with Konishi's study as well as to a separate study by Yang et al. [14] on the peripheral blood transcriptome of patients with IPF that identified DEFA3 to be increased in progressive disease. The apparent differences may be due to the different patient profiles and measures of disease severity. The AE-IPF cohort in Konishi's study had a mean % FVC of 55% and a lower DLCO (diffusing capacity of the lung for carbon monoxide) of 36%, and Yang's study used DLCO as the disease classifier.
Consistent with advanced disease, pathways involved in T-cell activation and cancer/tumour development were strongly enriched in the IPF molecular signature in our study. Notably, several checkpoint effector molecules including PD1 receptor, LAG3 and CTLA4 were shown to be upregulated in IPF tissue, indicating the possibility of immune exhaustion in end-stage IPF. The role of T-lymphocytes in human IPF and animal models of fibrosis has been recognised before [29]. Abnormal PD1 levels have been identified in human IPF, and human mesenchymal stem cell therapy was protective in a humanised mouse model of fibrosis through suppression of CD8+ T-cell infiltration and activation by a PD1/programmed death ligand (PD-L)1 axis [30]. It was recently reported that the increased expression of PD1 on CD4+ T-cells in IPF lung and plasma promotes a T helper (Th)-17 mediated response and induces active TGF-β secretion from stimulated T-cells [31]. Interestingly, the authors also show that knockout of PD1 as well as blockade of PD-L1 signalling ameliorates the fibrotic responses induced by bleomycin in mice. Our data emphasise the role of T-cell activation in advanced IPF and highlight the potential utility of this pathway as a disease classifier as well as a candidate therapeutic target. However, while the T-cell phenotype and lineage appear to be critically important in the context of IPF and particularly in severe IPF, more work is needed to understand their relative contribution to disease pathology.
While we noted increases in T-cell activation pathways, we also observed a reduction in genes involved in the activation of myeloid-derived suppressor cells and M2 macrophages (calgranulin, AGER and TLR4). Myeloid-derived suppressor cells are elevated in the peripheral blood of patients with IPF and inversely correlate with lung function [32]. Alternatively activated (M2) macrophages, described to be abundant in both murine and human fibrosis are known to drive anti-inflammatory and profibrotic responses simultaneously [33]. These macrophages also produce chemokines that recruit T-lymphocytes into the injured lung. Our observation of reduction in signals that activate M2 differentiation, together with an upregulation of chemokines involved in T-cell recruitment may imply that T-cell-mediated pathways contribute to late-stage disease.
Biopsies of early IPF are not in standard clinical practice and would be very difficult to procure in sufficient numbers to include in the study. Therefore, in our study design, we specifically included ALI as an additional comparator because both IPF and ALI represent severe lung pathologies, albeit with different aetiologies. Only cell adhesion and ECM remodelling pathways were commonly regulated in both these cohorts in comparison with non-disease controls. The IPF signature was rich in T-cell/immune response while the ALI signature was enriched in genes/pathways involved in cell cycle regulation. Thus, the IPF signature that we present here could be uniquely representative of end-stage lung fibrosis. Intriguingly, the cholesterol homeostasis pathway was significantly downregulated in our patients with advanced IPF with the signature overlapping with published statin-responsive genes in the lung [34], a finding that should be further explored and perhaps validated in independent cohorts. In addition, genes within lipoprotein steroid and fatty acid biosynthesis pathways were also strongly under-expressed in the IPF cohort. Given that lipid-lowering therapy and reduced lipoprotein levels have been shown to be profibrotic in preclinical and clinical studies [35, 36], our data lend further credence to the hypothesis that lipoprotein levels are inversely correlated with the pathogenesis of interstitial lung disease, although the mechanisms contributing to this effect remain to be investigated.
To date, there is only one study investigating alternative splicing in the IPF lung obtained through RNA sequencing of a small cohort of eight lungs from patients with IPF [27]. In that study, the authors used DEXseq to demonstrate alternative exon usage in known fibrosis-related genes including periostin and collagen 6. Our study identifies, for the first time, a larger subset of genes that exhibit alternative transcript/isoform usage in advanced IPF, opening up hitherto unknown targets and pathways for therapeutic intervention or as biomarkers of disease severity. Top pathways enriched in differentially spliced genes were found to be involved in fibrotic signalling (TGF, VEGF, ephrin B and LPA pathways) and cytoskeletal/ECM remodelling. Interestingly, we found HOPX (homeobox protein x), a known marker of alveolar Type I cells [37] as the top alternatively spliced gene in our study. A recent study showed that HOPX impacts alveolar epithelial injury and fibrosis and is decreased in human IPF tissue [38] contributing to lung function decline potentially through a failure of epithelial cell regeneration. Our expression data (downregulation of HOPX in IPF versus control) confirm these findings and suggest that alternative splicing of HOPX could play a role in the development of fibrosis.
We note that previously reported biomarkers (MMP7) and genes implicated in alveolar epithelial cell (DIO2) and surfactant functions (SP-A, SP-C) and lineage-negative lung progenitor cell expansion (KRT5) changed in total expression, validating previous findings [12, 39, 40]. However, exploring differential expression of exons can help identify genes that may not change in total expression but could be alternatively spliced in disease, a mechanism that is likely to be missed with microarray studies. For example, we identified Wnt2b, a molecule that is antifibrotic in hepatic stellate cells [41] as a major alternatively spliced gene although the overall expression was not changed in IPF tissue.
Prognostic and/or predictive biomarkers of disease progression in IPF are critical for the early diagnosis and treatment of the disease. In our cohort, we investigated the correlation of gene expression with clinical measures of disease severity, using % predicted FVC (recorded prior to transplantation) as the measure of lung function. Through Spearman's correlation analyses, we identified several genes whose expression was positively or negatively correlated with % predicted FVC. Many of the identified associations are novel. Interestingly, many do not appear to significantly change when comparing overall expression, but their expression level increased or decreased with disease severity. Our studies thus may have identified novel biomarkers of disease severity that can be validated in other independent cohorts.
In summary, we have described for the first time an RNA-Seq-based transcriptomic fingerprint of severe IPF and a molecular signature that can be further evaluated as a potential disease classifier. We chose not to directly compare this signature with previously described signatures from microarray platforms, as there are significant differences in patient disease profiles and the methods used to derive the molecular signatures. However, we note that as opposed to previous studies that have largely described epithelial injury, cell–matrix remodelling pathways, our cohort of patients with advanced IPF exemplify other unique pathways that could contribute to the disease and potentially present interesting targets and biomarkers. Recently, in an approach that complements ours, Reyfman et al. [11] used single-cell sequencing of advanced IPF lungs to generate a single-cell atlas of IPF and identify distinct subpopulations of alveolar macrophages and epithelial cells that could drive fibrosis. Similar to our observations, their work also identifies immune response, ECM organisation and Wnt signalling pathways as regulated in advanced fibrosis. Although we did not use a single-cell approach, which would have been cumbersome given the large number of samples involved, we believe that the whole-organ signature of advanced IPF is important and informative, since the disease phenotype in IPF is a collective consequence of the heterogeneity in cell types within the organ. RNA-Seq coupled with single-cell analyses could provide a new step forward in the elusive quest for biomarkers and targets for the diagnosis and treatment of IPF.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00117-2019.supp
Table T1 00117-2019.tableT1
Table T2 00117-2019.tableT2
Table T3 00117-2019.tableT3
Footnotes
This article has supplementary material available from openres.ersjournals.com
Conflict of interest: P. Sivakumar has nothing to disclose.
Conflict of interest: J.R. Thompson has nothing to disclose.
Conflict of interest: R. Ammar has nothing to disclose.
Conflict of interest: M. Porteous has nothing to disclose.
Conflict of interest: C. McCoubrey has nothing to disclose.
Conflict of interest: E. Cantu III has nothing to disclose.
Conflict of interest: K. Ravi has nothing to disclose.
Conflict of interest: Y. Zhang has nothing to disclose.
Conflict of interest: Y. Luo has nothing to disclose.
Conflict of interest: D. Streltsov has nothing to disclose.
Conflict of interest: M.F. Beers reports grants from Bristol-Myers Squibb during the conduct of the study.
Conflict of interest: G. Jarai has nothing to disclose.
Conflict of interest: J.D. Christie reports grants from the NIH and Bristol-Myers Squibb during the conduct of the study and grants from the NIH, GlaxoSmithKline and Bristol-Myers Squibb outside the submitted work.
- Received May 13, 2019.
- Accepted June 15, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










