





Abstract
Introduction The results of a clinical trial published in 2016 showed the efficacy of ivy leaves dry extract EA 575 versus placebo in the treatment of patients suffering from acute cough. A clinical trial with a very similar design was conducted to not only show the reproducibility of former results but also to investigate an alternative dosing scheme.
Methods This randomised, placebo-controlled, multicentre, double-blind clinical trial was conducted to assess the efficacy and safety of a liquid containing EA 575 in the treatment of acute bronchitis. A total of 209 patients were treated with a liquid containing EA 575 as an active investigational medicinal product (verum) either two (7.5 mL) or three (5 mL) times a day or placebo in the respective dosing scheme for 1 week, with a total observational period of 2 weeks. The primary efficacy outcome was a change in Bronchitis Severity Score (BSS) of the pooled placebo and pooled verum groups between visits 1 and 5. Additional secondary parameters were assessed, including, for example, change in cough severity as assessed by a visual analogue scale (VAS) and the Verbal Category Descriptive (VCD) score.
Results Superiority of verum over placebo was during and at the end of treatment, as measured by BSS. No significant differences between the dosing schemes were observed. VCD scores and VAS measurements also showed the superiority of verum over placebo.
Conclusion The existing data on the clinical efficacy of EA 575 were confirmed. Furthermore, a new dosing scheme was shown to be noninferior to the currently used scheme while maintaining the safety and tolerability of the well-established cough liquid containing EA 575.
Abstract
Ivy leaves dry extract EA 575 provides an effective and safe therapeutic option in the treatment of acute bronchitis http://bit.ly/318CZys
Introduction
Acute bronchitis usually presents a viral aetiology, is self-limiting and generally resolves within a couple of weeks [1, 2]. Its clinical symptoms vary greatly in duration and course, and they can change over the course of the disease [3]. The diagnosis of acute bronchitis is therefore essentially a clinical one, especially as there are no reliable diagnostic signs or laboratory tests [4]. Inflammation of the large airways of the lung, characterised by cough without pneumonia [1], can be considered a defining feature of an acute bronchitis. At the bedside, it is therefore typically diagnosed by physicians through cough in the absence of fever, tachycardia, tachypnoea or other clinical lung findings suggestive of pneumonia on examination [1, 5].
The treatment of cough, usually as a symptom of the common cold, has a long-standing history with plants that have been used since the early days of cultural history. Particularly in Europe, ivy (Hedera helix L.) has a long tradition in the treatment of bronchitis, where coughing is a significant symptom. While many medicines are being used today, undoubtedly not all therapeutics can fulfil the increased demand for high evidence of efficacy, the identification of the responsible active principles or the understanding of the mechanism of action. In this respect, the ivy leaves dry extract EA 575 stands out with over 20 published clinical trials and post-marketing studies, with documented clinical data with a total of >65 000 patients [6–8], in addition to preclinical data on the mode of action [9–11].
This degree of evidence obtained from EA 575 has led to the acceptance of ivy leaves dry extract as a valuable treatment option of inflammatory airway diseases [12]. There is a broad range of ivy preparations available on the market. However, many factors, including the manufacturing process of the extract, have an impact on its composition of (active) ingredients and thereby its beneficial properties [6]. Thus, different extracts, e.g. from ivy, are not interchangeable and the results of clinical trials with phytopharmaceuticals are not valid for the investigated plant(s) in general but only for the tested preparation [12].
We report the results of a randomised, placebo-controlled, double-blind, multicentre clinical trial to investigate the efficacy and safety of two dosing schemes of a liquid containing ivy leaves dry extract (EA 575 “verum”) against placebo in the treatment of acute bronchitis in adults. The results of this trial were recently presented in a poster [13]. The trial (EudraCT identifier number 2016-002426-37) was initiated after the completion and publication of a recently conducted, randomised controlled trial (RCT) (EudraCT No. 2014-003590-41 [7]) in order to increase the level of evidence while adding knowledge about the influence of a new dosing scheme for EA 575 treatment.
Methods
Study design
This phase III trial was conducted at five investigational sites: three general practices and two ear, nose and throat specialists in North Rhine-Westphalia, Germany. The aim of the study was to assess the efficacy and safety of a liquid containing EA 575 ivy leaves dry extract in the treatment of adults with acute bronchitis. The predetermined visits were defined as the initial visit V1 (day 0), V2 (day 2), V3 (day 3), V4 (day 4), V5 (7±1 days, end of treatment) and V6 (14±1 days, end of observational period).
Patients
The patient population in this trial consisted of male and female adults aged 18 to 73 years old, who were diagnosed with acute bronchitis and subsequently recruited from the patient pool of the investigators, based on clinical symptoms. Investigators assessed the symptoms (of which the lead symptom was cough) using the Bronchitis Severity Score (BSS), which is a published, validated tool to measure the severity of acute bronchitis [3, 14]. The patients had to have been symptomatic for 2–3 days before beginning the treatment and scoring ≥10 points in the BSS scale, ≥2 points on the Visual Category Descriptive (VCD) scale and ≥50 mm on a visual analogue scale (VAS) of cough severity. The most relevant exclusion criteria were allergic bronchial asthma, chronic or inherit lung diseases, and a history of chronic gastritis or peptic ulcers. Furthermore, treatment with corticoids, β2-sympathomimetics, other expectorants, antitussives, acetylsalicylic acid, nonsteroidal anti-inflammatory drugs, antibiotics, antihistamines or angiotensin-converting enzyme inhibitors 7 days prior to the first visit was prohibited. In addition, pregnant or nursing women, potential alcohol or drug abusers (as judged by the investigator), or patients with a body temperature >38.3°C were not admitted to the trial. Finally, investigators were asked to exclude the presence of any other serious disease that, in their opinion, could put the patient at increased risk by participating in the trial.
Treatment and randomisation
Patients that met all of the inclusion criteria and none of the exclusion criteria were randomised in a ratio of 2/2/1/1 (verum/verum/placebo/placebo) to receive active investigational medicinal product (IMP) treatment in the form of a daily dose of 105 mg EA 575 ivy leaves dry extract (drug/extract ratio (DER) 5–7.5/1 g) administered either via three doses of 5 mL per day or via two doses of 7.5 mL per day, or in the form of inactive IMP treatment consisting of the same two dosages of matching placebo. A randomisation list and sealed emergency envelopes were produced by an independent statistician, with a block size of six, reflecting the ratio of treatment arms (four verum and two placebo), using the software SAS (Statistical Analysis System; SAS Institute, Cary, NC, USA). At V1, each randomised patient received the IMP with lowest available randomisation number at the applicable site, which assigned the patient to one of the four treatment arms.
Engelhard Arzneimittel GmbH & Co. KG manufactured all IMPs in compliance with applicable Good Manufacturing Practice and Good Clinical Practice (GCP) guidelines. The 200-mL bottles containing IMP, either verum or placebo, were packed in identical outer cartons for each dosing scheme and identified with individual numbers according to the randomisation list. Only authorised personnel involved in the manufacturing process had access to the randomisation list, keeping it strictly confidential to all other involved parties. To keep the investigator blinded for the dosing scheme, the designated study personnel handled all IMP-related tasks. Monitoring of drug intake compliance was performed by weighing the IMPs at V1 and at V5. The use of IMP was calculated based on the difference in weight between those two values and compared with the calculated theoretical intake in order to evaluate the patient's compliance.
Concomitant medication allowed during the trial consisted of pharmaceutical contraceptives, paracetamol at a maximum dose of 2 g·day−1 but not 8 h before attending a visit and finally, of any other medication for concomitant diseases that was not prohibited by the exclusion criteria and was prescribed by a physician.
Investigational medicinal products
Independent of the dosing scheme, the active IMP (verum) consisted of a liquid containing ivy leaves dry extract (7 g·L−1 EA 575; extraction agent: ethanol 30% (m/m); DER 5–7.5/1 g), was well characterised [15] and quantified in the verum via its content of hederacoside C, with the final product being identical to Prospan Cough Liquid, which has a marketing authorisation in numerous countries. The placebo formulation consisted of the same components as the verum without the active ingredient EA 575. The flavouring and colouring system was adjusted in order to match the placebo in its final formulation to the flavour and colour of the verum, and thereby to obtain blinding.
Assessment of efficacy and safety
The primary efficacy variable was the change of the BSS between V1 (day 0) and V5 (day 7±1) with regard to the pooled verum treatments versus pooled placebo. The BSS is a sum score composed of the symptoms coughing, sputum, rattling noise, thorax pain and dyspnoea, each being rated on a point scale from 0 to 4 (0, absent; 1, mild; 2, moderate; 3, severe; 4, very severe), leading to a maximum value of 20 points. This score has been validated for the evaluation of bronchitis [14].
Furthermore, the following secondary efficacy endpoints were defined.
Difference in the BSS assessed over the whole observation period (between V1 and each of the visits V2–V6).
Difference in the BSS between V1 and V5 with regard to ivy leaves cough liquid versus placebo each in the dosage 3×5 mL.
Difference in the BSS between V1 and V5 with regard to ivy leaves cough liquid versus placebo each in the dosage 2×7.5 mL.
Difference in the BSS between V1 and V5 with regard to verum applied 3×5 mL versus verum applied 2×7.5 mL daily.
Cough severity assessed via VAS over the whole treatment period (area under the curve (AUC) over 7 days, V1–V5). Individual measurements were conducted by asking the patients to rate the severity of their cough on a horizontal line of 100 mm with anchors at 0 (no cough) and 100 (extreme cough) by a simple stroke of a pen.
Difference in cough severity assessed by the VCD score over the whole observation period (between V1 and each of the visits V2–V6) with the following rating options: 0, no cough; 1, one short period of mild cough without hardship; 2, some short periods of cough without much hardship; 3, frequent coughing that does not affect normal daily life or sleep; 4, serious coughing that is very frequent and interferes with normal daily life or sleep; 5, distressing continuous coughing that did not stop for 24 h.
Global efficacy assessment (GEA) at V5 and V6. The GEA consisted of answering two questions regarding efficacy, and was completed by the patients and the investigators using a five-point rating scale (0, very well; 1, well; 2, fair; 3, poor; 4, very poor).
Adverse events were documented from inclusion until V6, and the tolerability was assessed by the patient and the investigator at V5 and V6.
Statistical methods
Sample size calculation was based on the results of the trial EudraCT 2014-003590-41. For the primary variable change from baseline to V5 in BSS, a standardised difference of δ=0.81 was observed in the first trial [7]. With an allocation ratio of 2/1 for pooled active groups (n=120) versus pooled placebo groups (n=60), a power of >98% was calculated. For the comparisons of the active 5 mL treatment group (n=60) to the corresponding placebo group (n=30) and for the comparisons of the active 7.5 mL treatment group (n=60) and the corresponding placebo (n=30), the standardised difference was also δ=0.81, based on the results of EudraCT 2014-003590-41, with a two-sided α=5%. The corresponding power for both comparisons was calculated as 94%, with a noninferiority margin stipulated to −1.3 (i.e. 50% of the observed difference between verum and placebo in EudraCT 2014-003590-41) and a one-sided α=5%. ≥51 patients per treatment group were calculated to be needed for the comparison of the two vera. It was planned to enrol a total of up to 210 patients in order to cope with possible dropouts.
Continuous efficacy variables were summarised using the mean, standard deviation, median, and minimum and maximum values. Categorical variables were summarised using the cell frequencies and percentage of patients in each category. An a priori ordered hierarchical testing principle (closed test principle) to evaluate efficacy was used. The multiple level of significance was stipulated to α=5%. The hierarchical testing procedure controls the multiple significance level of 5%. The primary hypothesis H01 was: mean BSS change from baseline to V5 of pooled active groups=mean BSS change from baseline of pooled placebo groups. To show superiority of ivy leaves cough liquid, the primary hypothesis had to be rejected in favour of ivy leaves cough liquid, i.e. mean BSS change from baseline to V5 of pooled active groups had to be significantly greater than mean BSS change from baseline in the pooled placebo groups.
The following further ordered hypotheses were only tested confirmatorily in case of significance and this procedure controls the multiple α-level of 5%.
H02: mean BSS change from baseline to V5 of 5 mL verum group=mean BSS change from baseline of corresponding placebo group.
H03: mean BSS change from baseline to V5 of 7.5 mL verum group=mean BSS change from baseline of corresponding placebo group.
H04: mean BSS change from baseline to V5 of 5 mL active group is noninferior to mean BSS change from baseline of 7.5 mL active group. Noninferiority was concluded if the one-sided 95% confidence interval was within the region of noninferiority (−1–∞).
Ethical and legal aspects
The clinical trial was conducted according to International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) GCP, the principles of the Declaration of Helsinki, applicable regulatory requirements, standard operating procedures and the clinical trial protocol. A favourable opinion from the responsible Ethics Committee of the Ärztekammer Nordrhein (Duesseldorf, Germany) and approval from the German competent authority (BfArM, Bonn, Germany) were both received before recruitment of the first patient (ethics committee: 18 November 2016, trial registration number 2016277; BfArM: 18 August 2016, registration number 4041585). For all patients, written informed consent was obtained prior to trial inclusion.
Results
Of the 210 patients screened and enrolled, 209 patients were randomised and allocated to the full analysis set (FAS), and 208 took part until the completion of the trial at V6 (figure 1). Of the participants, a total of 49.3% were male and 50.7% were female. The average age was 36 years and participants were predominantly of Caucasian ethnic origin (table 1). The following results are based on the FAS. The baseline characteristics were evenly distributed among the subgroups.

Disposition of patients.
Demographics and baseline characteristics (full analysis set)
The primary efficacy outcome, the difference in change in the BSS from baseline to V5 between the pooled verum and pooled placebo groups, was met, with a mean difference in the respective BSS of 1.9 score points in favour of verum (p<0.0001) (table 2). Concerning secondary endpoints, statistical significance in favour of verum over placebo groups was reached on V3 (p=0.0002) (table 2). The individual dosing schemes (3×5 mL and 2×7.5 mL) likewise achieved statistical significance against the respective placebo group at V5 compared to baseline (p=0.0029 and p=0.0033, respectively). The comparison of the efficacy of both dosing schemes from baseline to V5 in turn showed no significant differences and, hence, the noninferiority (within the region of 95% CI –1–∞) of the tested 2×7.5 mL dosing scheme versus the dosing scheme in the authorised marketing registration of Prospan Cough Liquid (figure 2).
Development of Bronchitis Severity Score (BSS) over the whole observational period (full analysis set)

Mean Bronchitis Severity Score (BSS) over time (full analysis set). Data are present as mean±sd. #: p=0.0002; ¶: p<0.0001.
The secondary parameter relating to the cough severity was measured as the VAS of the pooled verum and placebo groups over the treatment period of 7 days (V1–V5), quantified by the AUC0–168 h. The AUC reflects the VAS after administration of drug over time. Lower values represent less severe cough. The results show superiority of verum (13% less AUC0–168 h) with statistical significance (p<0.0001). In a similar fashion, cough severity was assessed for both groups via the VCD score over the whole observational period of 14 days (figure 3). Statistically significant superiority of verum against placebo groups was achieved on V2 (day 2, p<0.0001) and was maintained until the end of the observational period on V6 (table 3), suggesting a continued treatment advantage even 1 week after treatment had ended.
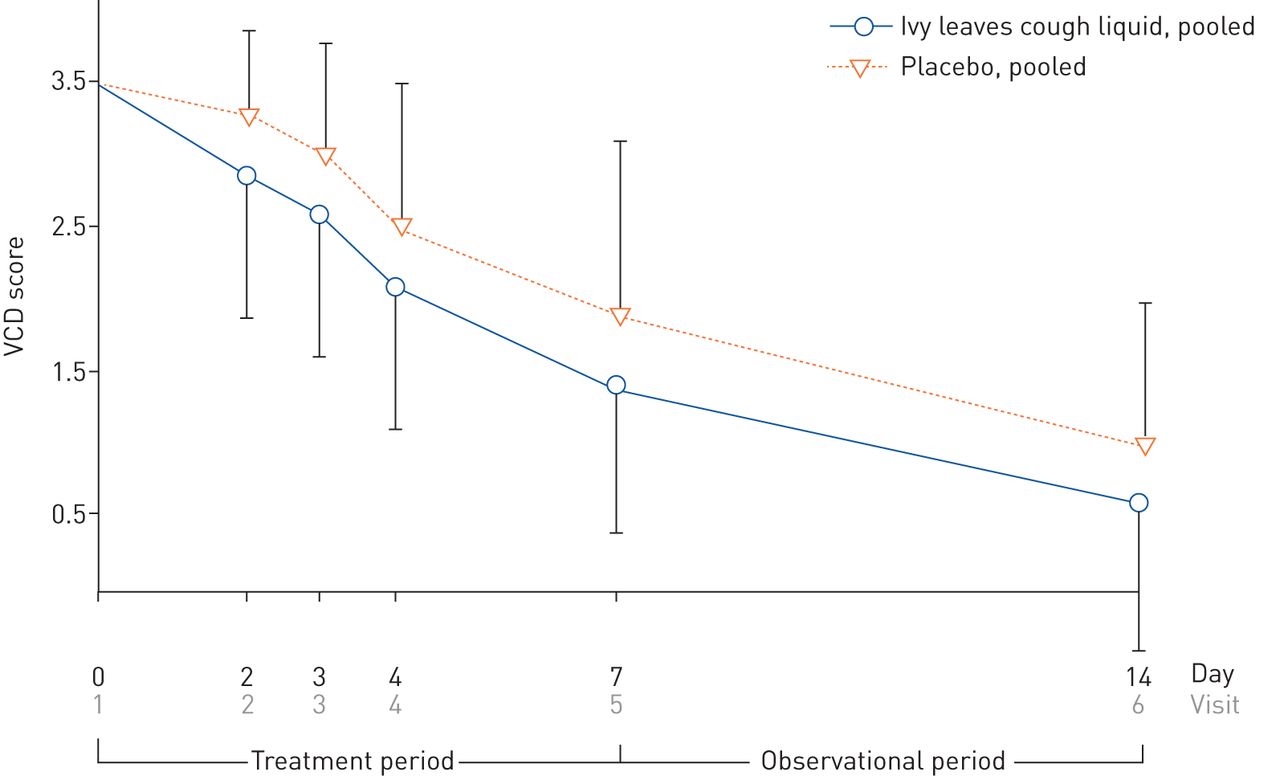
Development of Verbal Category Descriptive (VCD) score from visit V1 to V6 (full analysis set) of pooled verum and placebo groups. Data are present as mean±sd.
Development of Verbal Category Descriptive (VCD) score over the whole observational period
GEA was assessed at V5 and V6 (end of treatment and end of observational period respectively). Question A was: “Considering all the ways this treatment has affected you since you started in the clinical trial, how well are you doing?” Question B, on the other hand, was: “How do you rate this medication as a treatment for cough?” The assessments of the investigators did not differ markedly from the answers of the patients; hence, only the assessment of the patients is presented here. For question A, 79.1% of the patients regarded themselves to be doing well or very well in the pooled verum groups, contrasting with 50% giving the same answer in the pooled placebo groups at V5. At V6, in the pooled verum groups the results in the same criteria increased to 89.1% (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Results of the global efficacy assessment at visits a and c) V5 and b and d) V6 for questions a and b) A and c and d) B (full analysis set). Trend curves added for easy visualisation of main differences.
Concerning question B, 76.9% of the pooled verum groups rated the medication to be a good or very good treatment for cough, with 44.2% saying the same in the pooled placebo groups at V5. In summary, there were significant differences between both treatment groups regarding the assessments of both items on V5 and V6, respectively (p<0.0001), favouring ivy leaves cough liquid.
For most of the patients in both pooled treatment groups, the compliance was rated as “good” (>80% of the calculated theoretical intake had been applied). Only in one patient of the placebo group, was a “poor” compliance identified (weight of returned bottle showed that <50% of the total amount of trial medication had been applied).
Protocol deviations were discussed in the blind data review meeting and classified as “major” or “minor”. Moderate or poor compliance (15 patients) and violation of the inclusion criterion “symptoms of acute bronchitis lasting 48–72 h prior treatment” (five patients) were classified as major protocol deviations. Minor protocol deviations included deviations of the visit schedule and the patient who withdrew at V5, as all relevant assessments were completed.
During the entire observation period, 28 patients had at least one adverse event. A total of 29 adverse events (ivy leaves cough liquid: n=20 (14.4%); placebo: n=9 (12.9%)) were documented. 22 adverse events were attributable to a worsening of the cough severity measured by VAS based on the protocol requirement that worsening of >5 mm on VAS should be recorded as an adverse event. The adverse events were relatively well balanced between verum and placebo.
Beside the 22 adverse events that were evaluated as worsening of cough severity measured on VAS, a total of seven patients (ivy leaves cough liquid: n=6 (4.3%); placebo: n=1 (1.4%)) had one adverse event during the clinical trial. All adverse events in this clinical trial were nonserious and of mild or moderate severity, and for all the adverse events except for one, a causal relationship had been excluded. The only adverse events with a suspected causal relationship occurred in the ivy leaves cough liquid group (2×7.5 mL) and represented upper abdominal pain, which is expected according to the package insert of the marketed EA 575 cough liquid.
Tolerability was rated as “very well” by 66.9% and “good” by 32.4% in the verum groups at V5, and 74.9% and 25.5% respectively at V6. Patients who received placebo rated the tolerability as “very well” in 44.3% of the cases and “good” in 52.9% at V5, and 50.0% and 47.1% at V6. Assessments by the investigator did not differ in a relevant way from the patients.
Discussion
Despite the widespread occurrence of acute bronchitis, the number of publications offering current state of the art clinical data (RCTs and meta-analyses) on effective treatment options is marginal. Considering the enormous socioeconomic impact of acute bronchitis, corresponding treatment options are not only in high demand but also highly beneficial for society. This has recently been confirmed by the release of the updated guidelines for the diagnosis, and treatment of adults suffering from acute, subacute and chronic cough by the German Respiratory Society [12]. These guidelines highlight the aforementioned necessity of treatment by recommending the application of cough medications with evidence of efficacy through RCTs.
In a self-limiting disease such as acute bronchitis, a general improvement of symptoms is also expected for the placebo group. Thus, a proof of efficacy requires a significantly faster improvement of symptoms in the verum group. The primary endpoint, as measured via BSS, showed superiority over placebo at the end of the treatment period. Remarkably, there was still a significant treatment advantage at the end of the observational period 1 week after the end of treatment.
The number of patients included in this trial may be regarded as small to fully assess the clinical diversity of acute bronchitis in all its aspects. However, current guidelines on the development of clinical trials, such as ICH E9 (statistical principles for clinical trials) require a sample size calculation to be performed upfront in order to recruit as many patients as necessary to identify a statistically relevant outcome, while keeping the number of clinical trial participants as small as possible in order to reduce possible burden. In this respect, the number of patients included in this trial fulfils both objectives. The results of this trial corroborate the findings of a previous double-blind, placebo-controlled trial with a similar outcome [7]. A beneficial, clinically relevant effect of the treatment of the symptoms of acute bronchitis is therefore considered highly probable.
The fact that VAS values were not analysed per visit but only by using the AUC from baseline to the end of treatment might appear as a limitation of this trial. However, the applicability of this approach was verified in the previous trial, which was conducted with a similar design and the same IMP, by performing a statistical test on the correlation of VAS and BSS values. The results of the analysis showed a significant correlation between the VAS and the BSS total score at all visits indicating a similar profile for both variables over time.
It is also to be noted as a limitation that so far, no minimal clinically important difference has been published for the BSS and the investigated indication. However, the BSS is widely accepted and was validated in 2014 by Lehrl et al. [14].
The absence of an increase in adverse events at a higher single dose of 7.5 mL emphasises the safety of the active ingredient. Furthermore, the noninferiority of the twice-daily dosing scheme has the potential to improve patient adherence to the dosing scheme.
In conclusion, both formulations of the ivy leaves cough liquid were effective and safe in the treatment of acute bronchitis, and there were no differences between the two dosing schemes. The results confirm that the ivy leaves cough liquid has a favourable impact on the outcome of subjects suffering from acute bronchitis.
Acknowledgements
We would like to thank Alexander J. Pérez (Engelhard Arzneimittel GmbH & Co. KG) for writing support for this manuscript.
Footnotes
This study is registered at EudraCT with identifier number 2014-003590-41. The datasets generated during and analysed during the current study are available from the corresponding author on reasonable request, and will be published according to German Drug Law §42b.
Conflict of interest: A. Schaefer reports personal fees from Engelhard Arzneimittel GmbH & Co. KG.
Conflict of interest: F. Ludwig reports personal fees from Engelhard Arzneimittel GmbH & Co. KG during the conduct of the study and outside the submitted work, and she is an employee of Engelhard Arzneimittel GmbH & Co. KG.
Conflict of interest: B.M. Giannetti reports personal fees from Engelhard Arzneimittel GmbH & Co. KG.
Conflict of interest: M. Bulitta reports grants from Engelhard Arzneimittel GmbH & Co. KG.
Conflict of interest: A. Wacker reports personal fees from Engelhard Arzneimittel GmbH & Co. KG during the conduct of the study and outside the submitted work, and she is an employee of Engelhard Arzneimittel GmbH & Co. KG.
Support statement: All necessary funding was supplied by Engelhard Arzneimittel GmbH & Co. KG. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received January 16, 2019.
- Accepted October 8, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Licence 4.0.










