





Abstract
Nintedanib is a tyrosine kinase inhibitor approved for the treatment of idiopathic pulmonary fibrosis (IPF). In a retrospective, real-world study across seven Greek hospitals, we evaluated the effectiveness and safety of nintedanib in routine clinical practice. Patients diagnosed with IPF, as per guideline criteria or multidisciplinary diagnosis, received nintedanib between January 2013 and January 2018.
We evaluated 244 patients: mean±sd age 71.8±7.5 years, 79.1% male, 45.1% current smokers and 33.1% ex-smokers at treatment initiation. At baseline, predicted forced vital capacity (FVC) was 73.3±20.7% and predicted diffusing capacity of the lungs for carbon monoxide (DLCO) was 42.6±16.7%. On average, patients spent 23.6±15.0 months on nintedanib. At 3 years, 78 patients had died, equating to a 3-year survival rate of 59.4% (unaffected by treatment discontinuation or dose reduction). FVC% pred and DLCO% pred were largely stable at 3 years, with no significant difference from baseline (FVC 73.3±20.7% pred versus 78±20.1% pred, p=0.074; DLCO 42.6±16.7% pred versus 40.4±18.1% pred, p=0.334). Of the 244 patients, 55.7% reported an adverse event. Gastrointestinal events were the most common (173 (77.2%) out of 224 total events) and 45.0% of patients experienced diarrhoea. Only 32 (13.1%) patients had to permanently discontinue nintedanib due to an adverse event.
This real-world study shows a 3-year survival rate of 59.4% and a low discontinuation rate due to adverse events. Our experience is consistent with previous findings in clinical trials of nintedanib in IPF.
Abstract
Findings from the largest registry of Greek patients with IPF receiving nintedanib in routine clinical practice show, over a 3-year period, a low discontinuation rate and efficacy data that support the results of the INPULSIS clinical trials http://bit.ly/35a2CS5
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic interstitial lung disease, often with characteristic radiographic and histological features. Occurring primarily in older adults, IPF is commonly underdiagnosed [1]. The disease is associated with a high mortality rate; only two therapies (nintedanib and pirfenidone) are available to slow disease progression [2, 3] and these may be underprescribed [4]. Nintedanib (Boehringer Ingelheim, Ingelheim, Germany) was approved for the treatment of IPF based on the phase III INPULSIS trials, which involved 1066 patients from 24 countries [5, 6]. In the INPULSIS trials, nintedanib slowed IPF progression by reducing the annual rate of decline in lung function by 50% in a broad range of patient types, including patients with early disease (forced vital capacity (FVC) >90% predicted), limited radiographic fibrosis (no honeycombing) on high-resolution computed tomography (HRCT) and emphysema [7].
Although randomised controlled trials are initially necessary to characterise the efficacy and safety profile of a given drug, a fuller picture of that drug's effectiveness can be gained through real-world studies. One useful source of real-world data is registries, several of which have been established for IPF [8–14], and a growing number of real-world clinical experiences with nintedanib in IPF have now been published [15–21]. In the present study, we report data from the largest IPF registry in Greece to date, covering 244 patients treated with nintedanib for ≥3 years at referral respiratory departments across seven hospitals. We have included in our analysis data previously published in part by Tzouvelekis et al. [19] from 94 patients with IPF who received nintedanib between October 2014 and October 2016, substantially increasing the sample size of our study. The aim of this study was to assess the effectiveness and safety of nintedanib in routine clinical practice in Greece.
Methods
We conducted a multicentre, retrospective, observational, intention-to-treat (ITT) study across seven respiratory departments in Greek referral hospitals. We included 1) patients with a diagnosis of IPF according to criteria defined by collaborative guidelines published by the American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS) and Latin American Thoracic Society (ALAT) [22], and 2) patients with a confident diagnosis of IPF, as agreed by a multidisciplinary diagnosis discussion and based on a probable usual interstitial pattern (UIP) on HRCT scan in the correct clinical context [23]. All patients received at least one dose of nintedanib between January 2013 and January 2018. Data relating to investigator-reported adverse events, prior antifibrotic treatment, discontinuation and survival were recorded prospectively. Data from four centres were combined with extended follow up-data from the already published 1-year study by Tzouvelekis et al. [19] describing 94 patients receiving nintedanib, as well as the INPULSIS-ON extension study (seven patients) [24]. To avoid issues of potential duplicate publication, we state the following: 1) our current analysis involves a larger population of 244 subjects (compared with 94 subjects); 2) pooled safety and efficacy (functional) analysis of the combined cohorts was extended for a follow-up period of 3 years; and 3) additional outcomes are reported, including survival data.
Nintedanib was administered as per the manufacturer's recommendation (150 mg twice daily), and adverse events were managed through dose reduction to 100 mg twice daily [25]. Pulmonary function test (PFT) scores were taken at various time points, defined by the local clinical practice of each centre (patients were censored from the PFT analysis after death and/or discontinuation). The outcomes measured in this study were FVC%, diffusing capacity of the lungs for carbon monoxide (DLCO) % pred and mean survival after 3 years of nintedanib treatment. Lung function decline was defined as a reduction in FVC ≥10% pred. The institutional review board for human studies at each centre approved the protocol, and written consent was obtained from the subjects.
Statistical analysis
Statistical analysis was performed using SPSS 25 software (IBM, Armonk, NY, USA). Data are presented as mean±sd or median (interquartile range), as appropriate, for continuous variables, and frequency (percentage) for nominal variables. For the comparison of lung function change, a paired t-test was used, and for other subgroup comparisons, a one-way ANOVA or Mann–Whitney test was used. We used Kaplan–Meier analysis to assess survival, with p<0.05 the threshold of statistical significance.
Results
Patient characteristics
In total, 244 patients were treated with nintedanib in seven centres. Nintedanib was administered in three ways: 1) through the open-label extension trial of nintedanib (INPULSIS-ON) [24]; 2) under the manufacturer-funded named patient programme; or 3) through the Greek national health system. Most patients were male (79.1%) and had a mean age of 71.8±7.5 years. At study drug initiation, 45.1% of patients were current smokers, 33.1% were ex-smokers and 21.9% had never smoked. At baseline, mean FVC was 73.3±20.7% pred and mean DLCO% was 42.6±16.7% pred. Almost half of patients (43.8%) had advanced disease, defined as FVC ≤50% pred or DLCO ≤35% pred. Patients had a number of comorbidities at baseline, including chronic obstructive pulmonary disease (16.2%), coronary artery disease (31.3%), arterial hypertension (54.9%), atrial fibrillation (7.1%), gastroesophageal reflux (38.2%) and diabetes mellitus (22.0%). In addition, some patients developed lung cancer (7.3%) and pulmonary hypertension (26.4%) during the study period. Patients spent a mean 23.6±15.0 months on nintedanib (regardless of dose). Approximately one in 10 (11.8%) patients had previously received the antifibrotic treatment pirfenidone (Roche, Basel, Switzerland) for IPF. Baseline characteristics are shown in supplementary table E1.
Survival
In the observational ITT population, 78 patients died after 3 years of follow-up, equating to a 3-year survival rate of 59.4% (figure 1). Mean survival time was 54.7±3.5 months. Adjusting for discontinuation or treatment dose had no effect on survival (p=0.4 and p=0.7, respectively; see supplementary figures E1 and E2), although this was based on a small sample size. Survival data were not available for five patients.

Survival after 3 years on nintedanib treatment (n=239), mean±sd survival 54.7±3.5 months.
Lung function decline
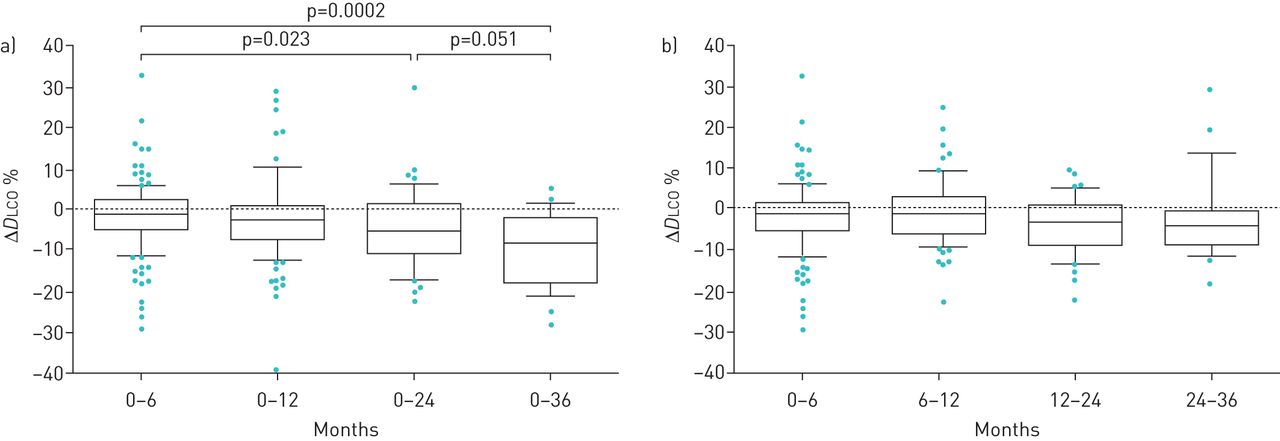
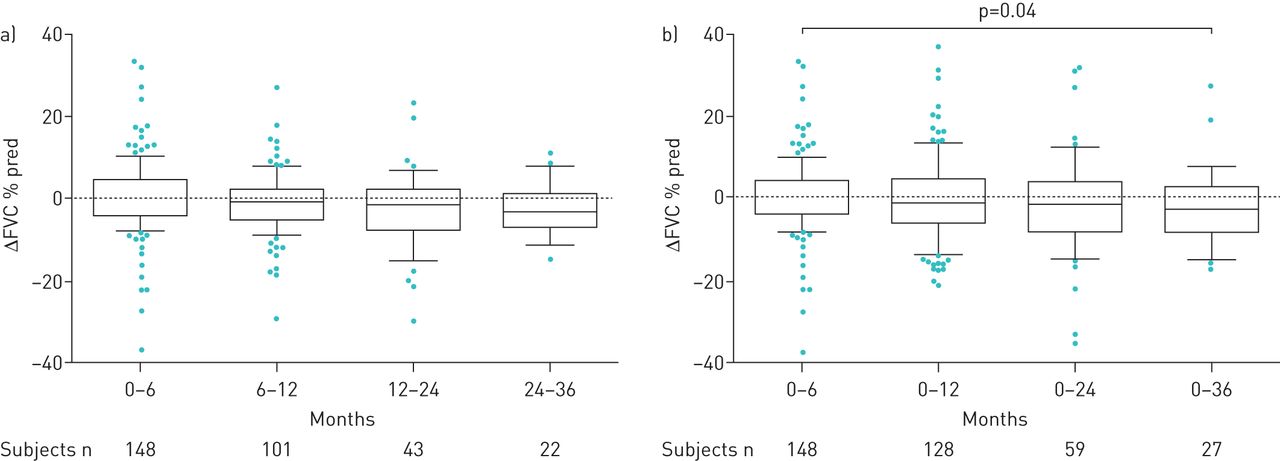
Mean±sd FVC% pred and DLCO% pred values at baseline, and then at 6, 12, 24 and 36 months after treatment initiation, are shown in table 1. We found no significant difference between FVC% pred at any time point compared with baseline (supplementary figure E3). However, FVC% pred increased between 12 and 24 months (74.6±23.4% pred versus 75.6±21.7% pred; p=0.028). An increase in FVC% pred occurred between 24 and 36 months of treatment, although this finding was not statistically significant (p=0.074; table 1).
Disease progression, as measured by percentage of predicted forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO) at 6, 12, 24 and 36 months after initiation of nintedanib in the studied population
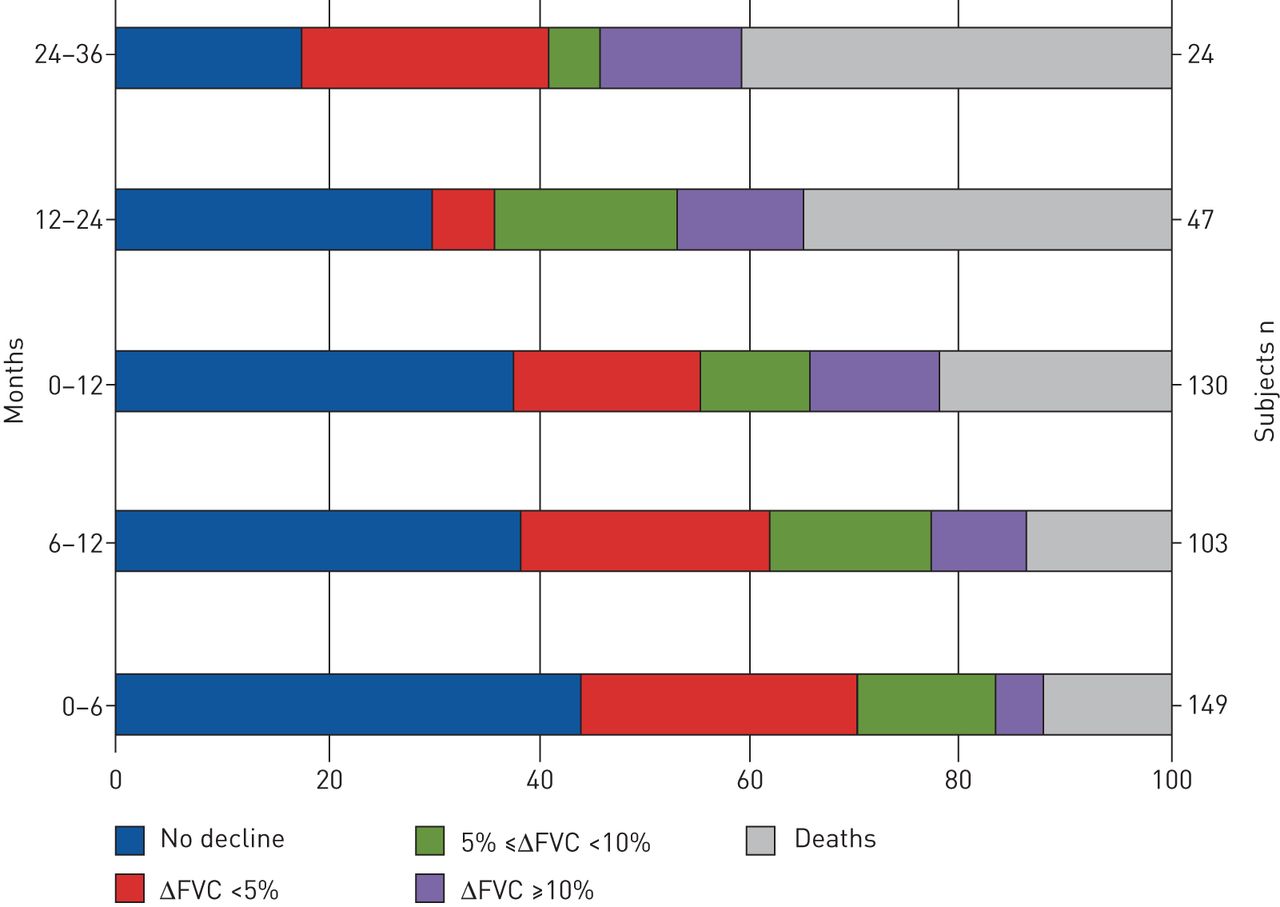
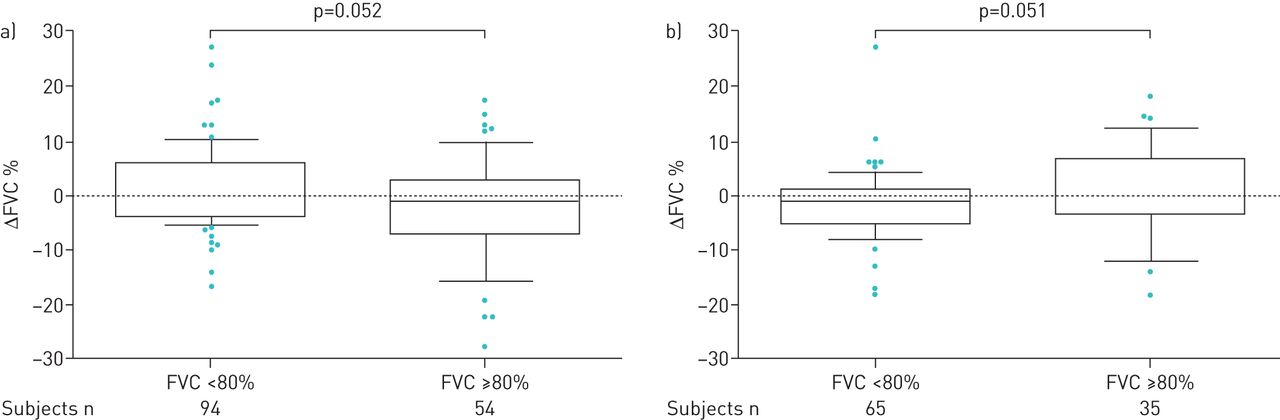
The mean values for decline in FVC% pred mirrored the findings in mean FVC% pred. We found no significant differences in mean change (Δ) in FVC% pred annually, although in the third year there was an increase in ΔFVC% pred (p=0.039; figure 2). Categorical FVC analysis during 3 years of follow-up showed that over the first year 35% of patients experienced progression (died or had FVC decline ≥10% pred). The proportion of patients that progressed between 12 and 24 months and 24 and 36 months expanded gradually, to reach 47% and 54%, respectively (figure 3). In addition, we performed a subgroup analysis based on FVC% pred at baseline (FVC <80% pred (n=151) versus FVC ≥80% pred (n=91)). In the first 6 months of treatment, and also between 6 and 12 months of treatment, there was a substantial yet borderline statistically significant difference between these groups (p=0.052 and p=0.051, respectively; figure 4). Comparisons between 0–12, 0–24 and 0–36 months were not significant (data not shown).

a) Change in forced vital capacity (FVC)% predicted at 0–6, 6–12, 12–24 and 24–36 months; b) change from baseline in FVC% pred at 6, 12, 24, and 36 months. Patients were censored from the analysis after death and/or discontinuation.

Proportion of patients with change (Δ) in forced vital capacity (FVC) <5%, ΔFVC ≥5–<10% and ΔFVC ≥10% at 6, 12, 24 and 36 months. Patients were censored from the analysis after death and/or discontinuation.
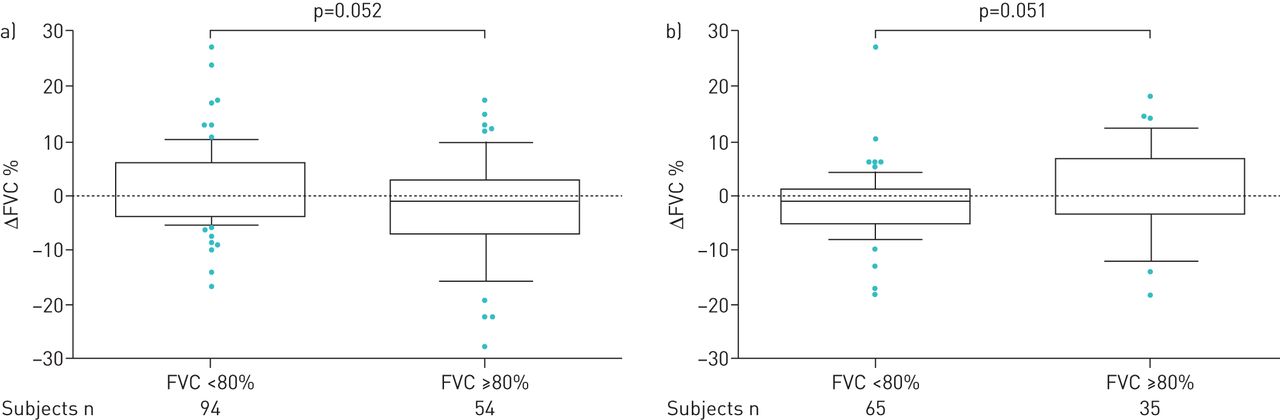
Change (Δ) in forced vital capacity (FVC) percentage between a) 0–6 months and b) 6–12 months stratified by baseline FVC (<80% (n=151) versus ≥80% (n=91)). Patients were censored from the analysis after death and/or discontinuation.
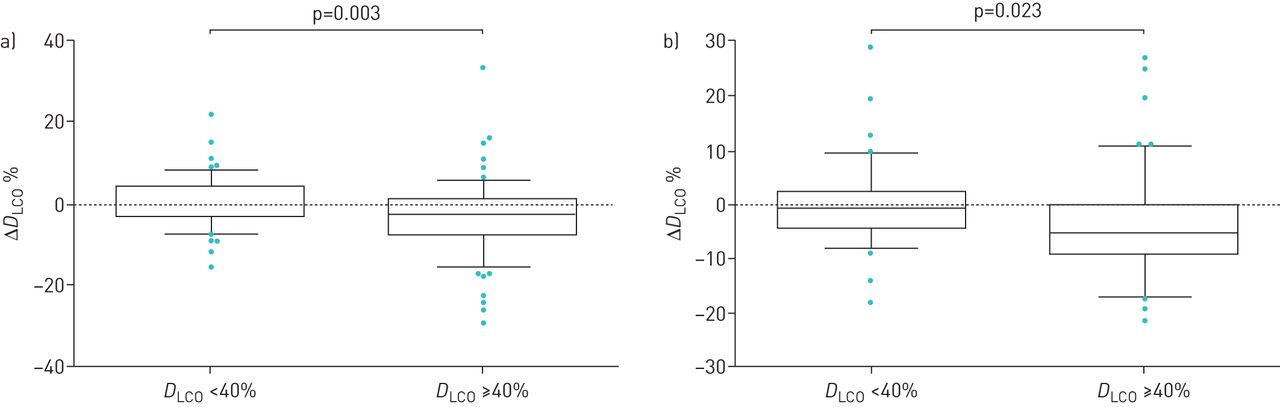
Mean±sd DLCO% pred was also stable during the 3-year follow-up period (supplementary figure E4). Although a significant difference was recorded in the first 6 months of follow-up, the mean±sd values do not suggest any clinical significance (table 1). A slight decrease in DLCO% pred occurred between 12 months (44.1±16.5%) and 24 months (42.1±14.6%) (p=0.002; table 1). Although the annual rate of DLCO percentage decline in the overall population remained relatively stable over the 3 years, there was a significant difference between the second and third years versus baseline (p=0.023 and p=0.0002, respectively; figure 5). When the data were stratified by baseline DLCO% pred (<40% versus ≥40%), we found that patients with DLCO% pred ≥40% at baseline had significantly higher rates of decline at 6 and 12 months versus patients with DLCO% pred <40% (p=0.003 and p=0.023, respectively; figure 6).
a) Change (Δ) from baseline in diffusing capacity of the lung for carbon monoxide (DLCO)% predicted at 6, 12, 24 and 36 months; b) change in DLCO% pred at 0–6, 6–12, 12–24 and 24–36 months. Patients were censored from the analysis after death and/or discontinuation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change (Δ) from baseline in diffusing capacity of the lungs for carbon monoxide (DLCO)% pred at a) 6 months and b) 12 months stratified by baseline DLCO (<40% (n=111) versus ≥40% (n=117)). Patients were censored from the analysis after death and/or discontinuation.
Safety
Only 32 (13.1%) patients had to permanently discontinue nintedanib due to an adverse event and 69 (28.3%) patients received a reduced dose to manage side-effects. Overall, 224 adverse events were reported in 136 (55.7%) patients, and the most common events were gastrointestinal (173 events; table 2). Of these, diarrhoea accounted for 110 events (45.0% of patients) and nausea/vomiting for 26 events (10.7% of patients).
Investigator-reported adverse events in the safety population
Discussion
We report findings from the largest IPF patient registry in Greece to date. For the first time, we present real-world, long-term efficacy and safety data for nintedanib from a group of Greek patients. The results of our analysis, which show a 59.4% survival rate, a low discontinuation rate and stabilisation of lung function during the 3-year follow-up period (as reflected in values for FVC% pred and DLCO% pred), are consistent with and add to the results of the INPULSIS-1 and INPULSIS-2 trials, in which nintedanib reduced FVC decline and slowed disease progression [5].
In the INPULSIS-ON extension study, 24% of patients who continued nintedanib and 27% of patients who initiated nintedanib died over ∼5 years of follow-up [24]. Pooled analysis using extrapolation estimated mean survival in six nintedanib trials to be 11.6 years for nintedanib-treated patients and 3.7 years for placebo-treated patients [26]. In the present study, 78 patients died in the 3-year follow-up period, equating to a 3-year survival rate of 59.4%. This discrepancy may be due to the fact that patients eligible for clinical trials tend to present with less severe disease and are more closely monitored than patients in the “real world”, such as those in registries, who may also have more comorbidities. While it is only possible to make an indirect comparison due to methodological differences, compared with the patients in the present study, those in the INPULSIS pooled nintedanib arm were younger (66.6 years versus 71.8 years), far fewer were current smokers (4.5% versus 45.1%) and their baseline PFT values were higher (e.g. FVC 79.7% pred versus 73.3% pred; DLCO 47.4% pred versus 42.6% pred) [27]. However, our results are comparable with the cumulative 3-year mortality rate of 37% observed in the Australian Idiopathic Pulmonary Fibrosis Registry [8]. Discontinuation had no effect on survival, although this could be attributed to the fact that most patients who discontinued due to adverse events received an alternative treatment.
Our study adds to the growing body of real-world evidence concerning nintedanib in IPF, which currently includes studies conducted in the UK, Germany, Italy and the USA [28]. After censoring the patients who died or discontinued therapy, our PFT analysis of FVC% pred over 3 years showed stable lung function and even slight improvement, in contrast to the significant rates of annual FVC decline observed in placebo-treated patients in IPF clinical trials [5, 29]. However, it is important to consider that patients who attended follow-up visits in our study could have had more stable disease than those who were lost to follow-up, which may account for the improvement in FVC observed between 24 and 36 months (despite not reaching statistical significance). This type of selection bias is inherent in all studies that use FVC as the primary outcome, and for this reason, overall survival may be a more useful end-point to consider.
The greatest decline in FVC% pred was in patients with higher preserved lung function at baseline, perhaps because they had more potential to decline. This is in contrast to the results of INPULSIS subgroup analyses, in which patients with preserved lung volume had the same rate of decline and derived the same benefit from nintedanib as patients with greater lung volume impairment [7, 30]. Identifying patients who may benefit from treatment is an emerging area of research, and subclinical interstitial lung abnormalities (ILAs) detected via HRCT offer a theoretical basis for the identification of early disease [31–33]. Since our patients were already diagnosed with IPF when they entered the registry, these data were not collected; however, we urge other clinicians collecting registry data and designing clinical trials to consider screening for ILAs in future.
It has recently been shown that many physicians in Europe are hesitant to initiate antifibrotic treatment [4], despite the plethora of safety data on antifibrotics. Although we did not record the severity of adverse events in this study, we adopted a practical, real-world approach to allow the maximum number of patients to continue on nintedanib. Wherever possible, we managed adverse events through dose reduction and advised discontinuation in severe cases only. As such, the rate of discontinuation in this study (13%) was lower than in INPULSIS-1 (21.0%) and INPULSIS-2 (18.8%) [5]. Furthermore, our observed discontinuation rates are lower than those previously published in other real-world studies in the UK (21% overall; permanent discontinuation n=30 (16%); temporary discontinuation n=9 (5%)) [15], Germany (40% overall; permanent discontinuation n=7 (11%); temporary discontinuation n=18 (29%)) [16] and the USA (26%) [17]. A time bias can often affect the reporting of adverse events; however, as patients who receive nintedanib typically report adverse events soon after the initiation of treatment, we do not believe a time bias affected our results.
We managed diarrhoea in the majority of our patients with diet recommendations, probiotics and dose reduction. Loperamide and/or codeine phosphate [34] were utilised in a minority of patients, who were predominantly older and with a higher rate of comorbidities, to avoid bowel-related functional obstructive symptoms. Reassuringly, the relatively low discontinuation rate in this study reinforces the results of the INPULSIS trials (albeit with a higher proportion of discontinuations (19.3%)) [27] to show that adverse events can be managed in most patients, using dose reduction when necessary.
The frequency of diarrhoea in our study (45.0%) is comparable with that observed in a UK (49.7%) study [15], but lower than that observed in other real-world studies, namely those in Germany (62.9%) [16] and the USA (52.6%) [17]. The reason for this discrepancy is not clear, but we speculate that patients in our study reported diarrhoea only when it was uncontrolled or disturbing enough for them to seek medical advice and management, hence the lower frequency. Equally, it may be that higher use of, or adherence to, natural remedies or anti-diarrhoeal medications contributed to the difference. It is interesting to note that lower frequencies of gastrointestinal adverse events associated with nintedanib have also been reported in other Mediterranean studies (53% in Spain and 40% in Italy) [35, 36], implicating the Mediterranean diet as a potential protective factor. Towards this end, a clinical trial is currently evaluating the effect of diet on the incidence of gastrointestinal adverse events in patients treated with pirfenidone (NCT03539289).
However, this real-world, retrospective evaluation does have its limitations. Some patients were lost to follow-up, meaning the number of patients with data after 3 years was reduced from the original ITT population. There is potential for this to cause bias, since patients with more advanced disease may be unable to attend follow-up visits and perform PFT re-evaluation. In addition, there are gaps in the data that would have provided a more complete picture of IPF outcomes in Greece, including acute exacerbations and causes of death. This is because data were collected from tertiary care centres in Greece, and not from the general practitioners involved in the patients' routine clinical care. It is also important to consider the homogeneity of the patient population, which included patients who were already receiving nintedanib, either through the open-label INPULSIS-ON extension study (n=7) [24], under the manufacturer-funded named patient programme, or through the Greek national health system. Homogeneity can sometimes limit the range of conclusions that can be drawn from a given population.
Another limitation is the lack of a control group. Because two antifibrotic drugs, nintedanib and pirfenidone, are now approved for the treatment of IPF, it was not possible in this study to compare against a placebo population, nor was it possible to compare against a historical cohort treated with alternative agents (prior to the availability of antifibrotics). Lastly, we included patients with a probable UIP pattern identified by HRCT scan and a definite diagnosis of IPF as per the Fleischner Society criteria. According to the most recent ERS/ATS/JRS/ALAT guidelines [22], these are not sufficient to make a confident diagnosis of IPF. However, in support of our inclusion criteria, the evidence suggests that in most cases, a possible UIP pattern with a consistent clinical background predicts a definite UIP on surgical lung biopsy [37, 38].
Data from extension studies and real-world studies should always be interpreted with caution [39]. Nevertheless, we believe that this study provides useful real-world evidence on the efficacy and safety of nintedanib, without some of the selection biases inherent in clinical trials. Our real-world efficacy and safety data are aligned with those from tightly controlled clinical trials, which is reassuring for both patients and respiratory physicians. To further characterise the optimum approach to dose reduction and discontinuation, continued monitoring of the effectiveness of nintedanib in routine clinical practice is warranted.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00172-2019.supplement
Acknowledgements
We thank the patients who participated in this study and their families. We also thank Thodoris Karampitsakos (First Academic Respiratory Dept, University of Athens, Greece) for his substantial contribution to the collection and analysis of study data.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Author contributions: The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. The authors received no direct compensation related to the development of the manuscript. Writing support was provided by Chester Trinick of MediTech Media (London, UK), a medical communications agency contracted and funded by Boehringer Ingelheim International GmbH (BI). BI was given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations. All authors were involved in the conception and design and drafting of the manuscript, and review and revision of the manuscript for important intellectual content. Katerina Antoniou, Argyrios Tzouvelekis, Katerina Markopoulou, Athina Trachalaki, Eirini Vasarmidi and Jiannis Organtzis contributed equally.
Support statement: Medical writing assistance was supported financially by Boehringer Ingelheim. Some data reported in this manuscript are derived from the Investigating Idiopathic Pulmonary Fibrosis in Greece (INDULGE IPF) registry, supported financially by Boehringer Ingelheim.
Conflict of interest: K. Antoniou reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work.
Conflict of interest: K. Markopoulou reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work.
Conflict of interest: A. Tzouvelekis reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work. In addition, Dr Tzouvelekis has a patent on inhaled or aerosolised delivery of thyroid hormone to the lung as a novel therapeutic agent in fibrotic lung diseases issued to Yale University.
Conflict of interest: A. Trachalaki has nothing to disclose.
Conflict of interest: E. Vasarmidi has nothing to disclose.
Conflict of interest: J. Organtzis has nothing to disclose.
Conflict of interest: V. Tzilas has nothing to disclose.
Conflict of interest: E. Bouros has nothing to disclose.
Conflict of interest: G. Kounti reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work.
Conflict of interest: C. Rampiadou reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work.
Conflict of interest: S-C. Kotoulas has nothing to disclose.
Conflict of interest: F. Bardaka has nothing to disclose.
Conflict of interest: E. Bibaki has nothing to disclose.
Conflict of interest: E. Fouka reports travel grants, lecture fees and consultation fees from Boehringer Ingelheim and GlaxoSmithKline, and travel grants and lecture fees from Roche Pharma, AstraZeneca, Novartis, Chiesi, Innovis and Elpen outside the submitted work.
Conflict of interest: G. Meletis reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study.
Conflict of interest: S. Tryfon has nothing to disclose.
Conflict of interest: Z. Daniil reports grants, personal fees, nonfinancial support and other support from BI Hellas during the conduct of the study, and other from Roche outside the submitted work.
Conflict of interest: D. Papakosta reports financial support for research, travel grants and lecture fees from Boehringer Ingelheim and Roche, outside the submitted work.
Conflict of interest: D. Bouros reports research support, personal fees for lectures and advisory boards, and travel grants from Boehringer Ingelheim, and personal fees for lectures and advisory boards, and travel grants from Roche, outside the submitted work.
- Received July 8, 2019.
- Accepted November 13, 2019.
- Copyright ©ERS 2020
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










