





Abstract
Inflammatory myofibroblastic tumours (IMT) are a rare cause of endobronchial masses in adults. Surgery has been the mainstay of treatment of endobronchial IMTs, based on the potential for recurrence. Interventional pulmonology has emerged as a minimally invasive and lung function preserving modality in management of airway obstruction due to tumours. We present a series of three adult patients with IMT treated endobronchially with a short discussion on its potential role. We also discuss how molecular analysis of IMTs for mutations in genes such as ALK and ROS1 might provide insights into clinical behaviour and potential targetable therapy in advanced, unresectable and metastatic cases.
Abstract
Inflammatory myofibroblastic tumours in central airways: bronchoscopic treatment of a “not so pseudo-” tumour https://bit.ly/2KgPntq
Introduction
Inflammatory myofibroblastic tumour (IMT) is a rare mesenchymal tumour that presents as a pulmonary or soft tissue mass, mostly in children and young adults with an intermediate potential for recurrence, rarely local invasion and metastasis [1]. It is a neoplastic proliferation of myofibroblasts always accompanied by non-neoplastic inflammatory cells such as lymphocytes, plasma cells or macrophages. Recent insights in the molecular genetics of these tumours allow for a more precise delineation from the non-malignant mimickers [2].
Endobronchial IMTs are rare in adults. Most available literature on bronchoscopic resection of IMTs pertains to tumours in the paediatric age group. We present three cases of endobronchial IMTs resected with bronchoscopic techniques along with a review of literature on the same. To the best of our knowledge, this is the first such series in adults. In addition, we discuss newer insights into the molecular genetics of the tumour and treatment modalities for advanced and metastatic lesions.
Case 1
A 42-year-old man presented with increasing dyspnoea, wheezing and haemoptysis. He had presented with right testicular carcinoma 2 years before, which was treated with surgical resection and adjuvant chemotherapy. Computed tomography showed a mass in the right main bronchus, and possibility of metastasis was considered.
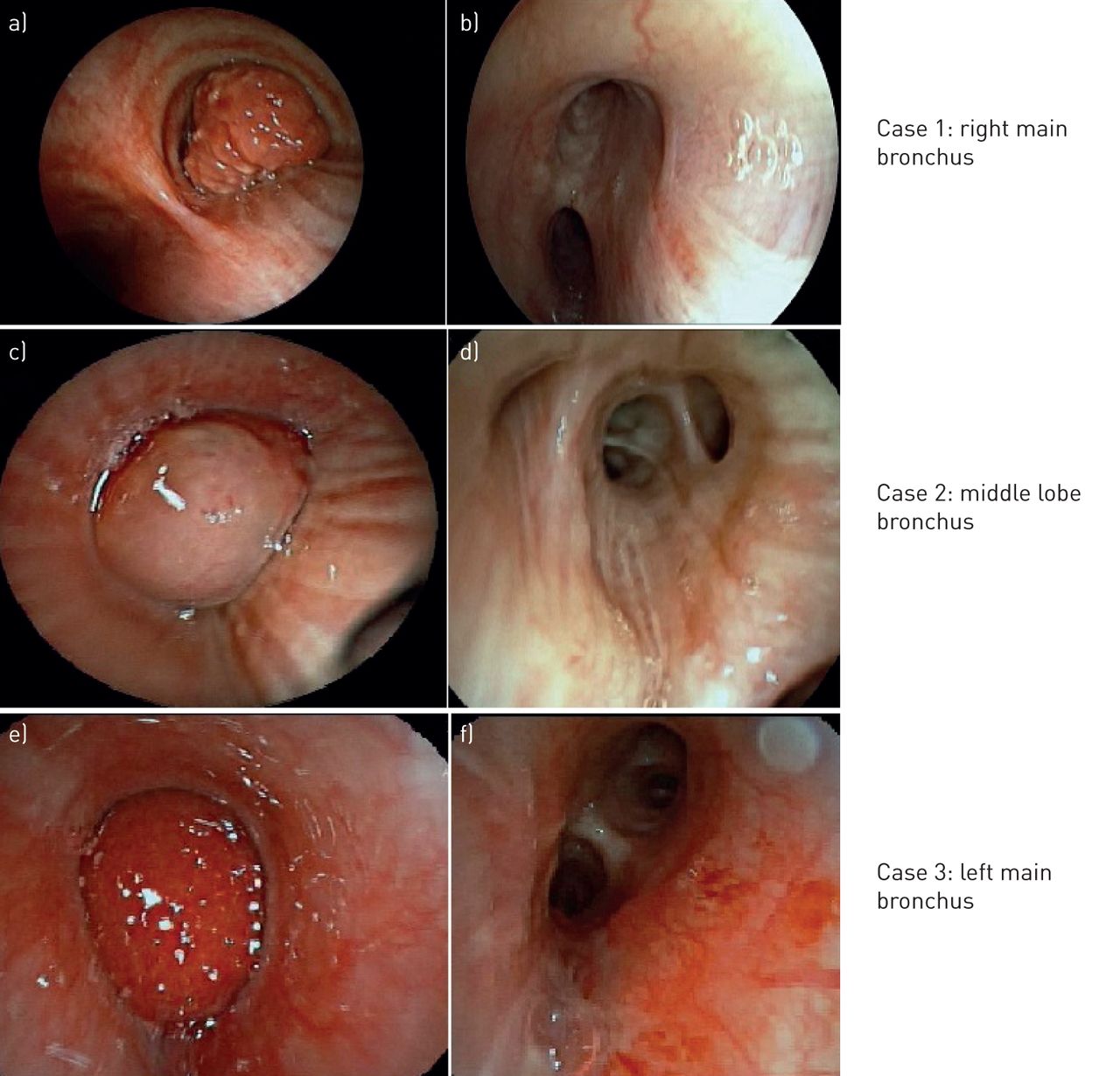
Urgent rigid bronchoscopy revealed a globular tumour blocking 90% of the right main bronchus (figure 1a). The tumour was completely resected with the diathermy snare and the tumour bed was treated with diathermy.

Bronchoscopic view a, c, e) before and b, d, f) after bronchoscopic resection of inflammatory myofibroblastic tumours.
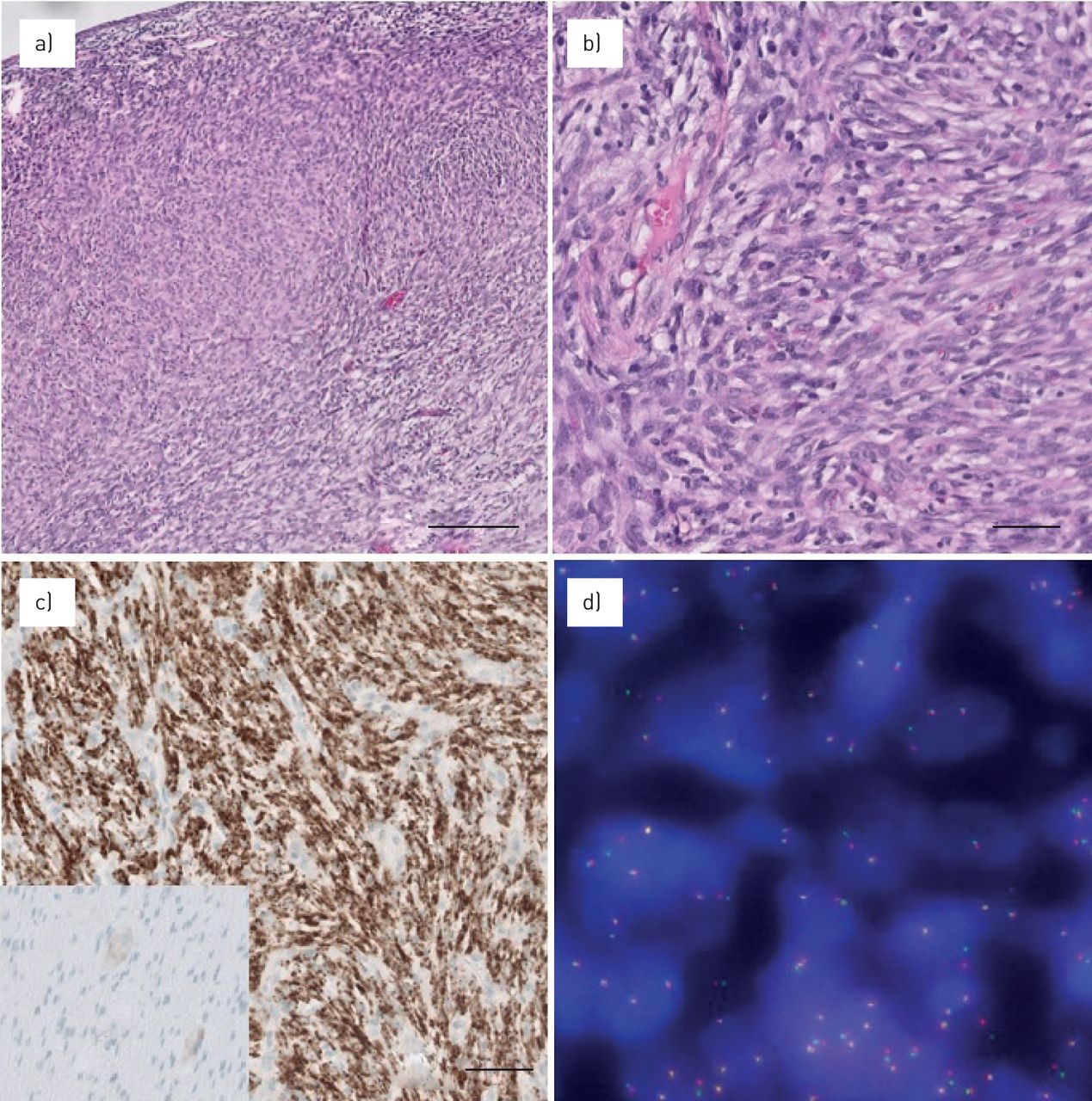
Histological examination showed spindle shaped cells with prominent nucleoli and eosinophilic cytoplasm arranged in short fascicles with occasional mitoses in the tumour cells (figure 2a and b). The stroma was infiltrated with lymphocytes, histiocytes and occasional eosinophilic granulocytes. On immunohistochemistry (IHC), tumour cells stained positive for ALK-p80 (figure 2c). ALK fluorescence in situ hybridisation (FISH) was positive with split signals. FISH revealed a rearrangement in ALK (figure 2d), and hybrid capture based next generation sequencing (NEOselect) showed EML4-ALK1 fusion. Diagnosis of inflammatory myofibroblastic tumour was made.
{kind=link}
{kind=link}
A representative overview and detail of inflammatory myofibroblastic tumour is shown. Haematoxylin and eosin stain a) 10× and b) 40×. c) Immunohistochemistry is positive in tumour cells for ALK (5A4 antibody, 20x), including the control appendix tissue showing negative myocytes and weakly positive ganglion cells (frame within panel (c)). d) Fluorescence in situ hybridisation analysis with an ALK rearrangement, note the split signals.
Bronchoscopy after 1 month showed necrotic tissue at the tumour base. Follow-up with bronchoscopy and computed tomography has revealed no recurrence in 38 months (figure 1b).
Case 2
A 49-year-old man presented with haemoptysis and breathlessness of 1-month duration. Computed tomography of the chest showed a mass in the middle lobe bronchus with atelectasis of the middle lobe.
Urgent bronchoscopic intervention was planned due to increasing dyspnoea. A flexible bronchoscope was passed through the channel of the rigid bronchoscope. A smooth rounded tumour covering the right middle and lower lobe bronchi was seen (figure 1c). The tumour was removed with the diathermy snare and the tumour base at middle lobe carina (RC2) was treated with diathermy. Bleeding was controlled with endobronchial instillation of epinephrine, tranexamic acid and use of a bronchial blocker.
Histology showed epitheloid cells with granular cytoplasm arranged in short fascicles with infiltration of lymphocytes. Tumour cells were negative for S100 on IHC, ruling out a granular cell tumour. Subsequent ALK-p80 was positive. FISH- and hybrid capture-based next generation sequencing confirmed the diagnosis of EML4-ALK1 fusion-positive IMT.
Rigid bronchoscopy was repeated after a month. Abnormal mucosa on the roof of right middle lobe bronchus and RC2 was treated with diathermy and cryotherapy. Biopsies showed no residue. Bronchoscopy at 5 months showed an oedematous right middle lobe opening, but biopsies were clear of residual tumour. Follow-up with computed tomography scan and bronchoscopy showed no recurrence in 40 months (figure 1d).
Case 3
A 16-year-old girl presented with cough and was diagnosed as having pneumonia that was treated with antibiotics. 1 month later she presented with pneumomediastinum. Computed tomography scan showed a tumour in the distal left main bronchus with atelectasis of the left lower lobe.
Rigid bronchoscopy revealed a tumor in the distal left main bronchus, vascular and bleeding easily on touch. The surface of the tumor was coagulated with cautery and mechanical debulking with the rigid scope was done (figure 1e and f). Bleeding from the tumour bed at the ventro-medial aspect of distal left main bronchus was controlled with diathermy and pressure with a bronchial blocker. The procedure was stopped at this point and the patient was advised a follow-up.
Histology showed spindle shaped tumour cells with elongated nuclei, prominent nucleoli and eosinophilic cytoplasm arranged in a herring bone pattern, with infiltrates containing lymphocytes and occasional eosinophilic granulocytes. The cells stained positive forALK-P80 on IHC, FISH- and hybrid capture-based next generation sequencing showed an EML4-ALK1 fusion positive IMT.
After 2 months, repeat bronchoscopic treatment for residual tumour with application of diathermy was done. Biopsies after 4 months were clear of tumour. After 7 months, a recurrence was noted at the distal left main bronchus that was treated with cryotherapy and diathermy. Biopsies at 9 and 12 months were clear of tumour. After 18 months, computed tomography showed recurrence in the distal left main bronchus and bronchoscopic biopsies confirmed ALK positive IMT. The patient was referred for sleeve resection of the distal left main bronchus and surgical resection was achieved with margins being clear of tumour. In the resected tumour, the EML4-ALK1 fusion was confirmed. FISH for ROS1 and RET mutations were negative. The patient was asymptomatic and computed tomography scan 30 months after surgery has shown no evidence of recurrence.
Discussion
IMT is a soft tissue neoplasm of mesenchymal origin composed of spindle cell myofibroblasts accompanied by an inflammatory reaction. Nomenclature of IMT was ambiguous in the past. Terms such as inflammatory pseudotumour (IPT), plasma cell granuloma, pseudoxanthomatous tumours, were used interchangeably and it was considered to be benign. Subsequently, it was noted that a subset of these pseudotumours had clonal proliferation, harboured genetic mutations and showed potential for local recurrence and distant metastases. These tumours were recognised as IMTs and classified as an intermediate grade bone and soft tissue neoplasm by WHO [1–3]. Approximately 50–70% of IMTs harbour rearrangements of ALK-4, ROS1, PDGFRβ, RET and ETV6 genes, most of them members of the mitogen-activated protein kinase pathway [4, 5]. These insights into the molecular genetics of IMT denote a malignant potential and delineate it from benign mimickers like immunoglobulin (Ig)G4-related plasma cell granulomas, inflammatory pseudotumour of the lymph node and spleen or mycobacterial pseudotumour. These genetic alterations result in activation of the tyrosine kinase receptor and may offer novel possibilities for treatment with receptor kinase inhibitors in unresectable and metastatic cases [6, 7].
IMTs can involve many organs including abdomen, mesentery, lung, head, neck, extremities, genito-urinary tract and orbit. Local recurrence rates of 15–37% and metastases rates of 5–11% have been reported [2, 3, 8].
Incidence of pulmonary IMT in adults is around 0.04–1% of all lung tumours. Most tumours are found in patients less than 40 years of age with a reported mean range of 27–50 years. No gender predilection has been noted. Endobronchial growth of IMT is rare, with a prevalence of 0–12%, although one study reported 22% [9].
Tracheobronchial IMTs may present with airway obstruction, respiratory distress or collapse of the distal lung. Rare presentations include pneumothorax [10] and pneumomediastinum as noted in one of our patients.
Surgery is often the first treatment choice but may result in significant loss of lung function especially if the location of the tumour renders it unsuitable for parenchyma sparing surgeries such as sleeve resection. Bronchoscopic removal is a minimally invasive technique and a valuable tool for preserving lung function especially if the tumour is purely endoluminal [11, 12]. Bronchoscopic removal of IMTs has been well documented in the paediatric age group and recurrence was mainly noted following partial resection. In adults, data are largely restricted to case reports.
We conducted a review from PubMed, Embase and Medline database for case reports and series wherein bronchoscopic methods were used as an intervention for management of inflammatory myofibroblastic tumours. Studies with full text available in English were included in the analysis.
We reviewed 27 articles that included 22 case reports and five case series [10–36].
Including the three patients from our series, 37 patients underwent resection of endobronchial IMT with the help of bronchoscopic techniques. 22 (69%) patients were over 16 years of age. The most common site of tumour involvement was the left main bronchus (17) followed by the trachea (14). 24 (65%) out of 37 patients were successfully treated with bronchoscopic interventions. Modalities of therapy included use of the rigid bronchoscope with mechanical or forceps debulking (17), laser resection (10), use of diathermy (9), argon plasma coagulation, cryotherapy and electrocautery snare. Flexible bronchoscopic resection was undertaken in one patient.
Five patients had to undergo repeat bronchoscopic procedures, following which the tumour was completely removed [12, 26, 29, 30].
Surgery was done after primary bronchoscopic treatment in 12 patients. Indications for surgery were elective (3) [10, 14, 29], incomplete bronchoscopic resection (2) [15, 21] extraluminal component (1) [25] and recurrence (6) [18, 23, 27, 28, 32]. One patient was deemed too old and unfit for surgery due to severe COPD and hence endobronchial treatment was palliative [19]. Thus, only eight (21%) of the 37 patients who underwent bronchoscopic resection as the primary treatment modality needed surgery for tumour recurrences or incomplete removal. Mean follow-up period after successful resection was 22 months and mean time to recurrence, when noted, was 14 months.
Considering the rare occurrence of this tumour, it is unlikely that a prospective study comparing success rates of surgery versus endoscopic removal will be conducted.
Endoscopic resection can also be a useful approach in conditions such as pregnancy, for which the risk of definitive surgery may be higher [37] or in patients who are unfit or unwilling for surgical resection [19, 36]. Even with large tumours having extra-luminal components, it is useful in managing respiratory distress as a bridge until the patient is fit to undergo surgery [14, 21, 25]. This approach was used for 10 out of the 11 paediatric patients with IMTs who subsequently underwent definitive surgery for submucosal components [38].
When single step resection is not possible, a repeat procedure may also be successfully performed [12, 26, 29, 30] as was the scenario in five cases including one from our series. Regular follow-up with computed tomography scans and repeat bronchoscopy with biopsies when required is mandatory to assess response and recurrences.
Endoluminal location of the tumour may be confirmed with imaging with thin slice computed tomography scan and endoscopic evaluation. Radial endobronchial ultrasound (EBUS) can be used to determine if the tumour is extending beyond the cartilage or is limited to submucosal layers and is superior to computed tomography scan in this respect. For better approximation in proximal airways the central probes with balloon sheath may be used for airway assessment. The normal central airways have seven layers visualised on radial EBUS: the innermost two are the mucosa and submucosa, the third, fourth and fifth correspond to the cartilage, and the external two are loose and dense fibroelastic tissue surrounding the airway. One of the first indications for use of EBUS was for identifying extent of tumour extension prior to endobronchial treatment of tumours. In differentiating tumour compression and infiltration, EBUS has an accuracy of 94% [39]. Linear EBUS may be used to detect nodal involvement. IMTs may be FDG avid and positron emission tomography computed tomography may be a sensitive tool in determining pulmonary and mediastinal involvement, distant metastases, recurrences and residual disease [40]. Advances such as confocal laser endomicroscopy and biopsy may be an added asset in assessing adequate resection and possible recurrences in the future [41].
An important aspect to be considered is the vascularity of the tumour and the potential for bleeding. Significant bleeding was noted in two of our cases requiring use of haemostatic measures including the use of a bronchial blocker. It is imperative that resections be undertaken at centres with the required expertise and facilities for haemorrhage control.
On histology, IMTs show the presence of spindle cells of myofibroblast origin with low mitotic activity, an inflammatory component of plasma cells, lymphocytes and histiocytes, and fibromyxoid stroma. The ratio of different components may vary. Some cases show a prominent number of inflammatory cells, while others may have a mild inflammatory component. The cytoplasm of the spindle cells may be homogeneously stained or sometimes appear granular. Histological spectrum is quite broad and three patterns have been described, as follows [42]. 1) Myxoid/vascular which resembles nodular fasciitis with loosely arranged plump spindle cells in a myxoid stroma and prominent vasculature. This variant may have more eosinophils or neutrophils and fewer plasma cells than the other types. 2) The compact spindle cell pattern with a cellular proliferation of spindle cells and a fascicular architecture in a collagenous stroma. These foci show numerous plasma cells and lymphocytes. 3) Fibromatosis type which is relatively hypocellular with elongated spindle cells in a densely collagenous background with lymphocytes, plasma cells and eosinophils.
The differential diagnosis includes reactive lesions like granulation tissue and plasma cell granulomas [1–3]. Differentiation from reactive lesions can be difficult on morphology alone. High cellularity, and the fascicular, relatively monomorphic growth pattern of the IMT can be helpful in this matter. Standard immunohistochemistry is mostly of no specific help, SMA is positive as it is in the reactive myofibroblasts. However, molecular abnormalities, such as ALK, ROS1, RET and NTRK1 fusions are definite in the differentiation from reactive pseudotumours. If immunohistochemistry is used for fusion screening, a positive ALK/ROS1 should always be confirmed by an orthogonal technique, FISH or next generation sequencing as we presented in our cases.
In the second case of our series, a granular cell tumour was considered in the differential diagnosis due to prominent granular cytoplasm. The absence of S100 immunohistochemistry staining renders a granular cell tumour unlikely.
Nuclear pleomorphism and atypia are rare in the myofibroblasts of IMT, and when present should warrant that a differential of sarcoma should be ruled out.
Clonal cytogenetic rearrangements that activate the ALK receptor kinase gene in the chromosome band 2p23 emerged in recent years as genetic drivers in IMTs harbouring different fusion partners. Immuno-histochemistry for ALK1 may be positive in 45–50% of IMTs and shows a granular or perinuclear staining pattern. In ALK-negative cases other rearrangements may be found. Its genetic profile closely resembles that of lung adenocarcinoma even when it arises in soft tissue and other organs. In 10% of IMTs, ROS1 rearrangement was found. Occasionally tumours harboured RET, PDGFRβ and ETV6 gene rearrangements [5, 6]. These new findings imply that IMT is a kinase fusion-driven neoplasm [4]. Interestingly, around 90% of paediatric IMTs are ALK fusion positive whereas only 10% of adult IMTs are ALK fusion positive tumours [5]. Roughly 30% of IMTs do not harbour any known mutations or genetic rearrangements, mostly in adults. Novel genes might still emerge as drivers of this neoplasm. The presence of ALK- RANBNP2 fusion has been associated with a distinct variant of IMT with round or epithelioid cells on morphology, a perinuclear staining pattern and an aggressive clinical course. The term epithelioid inflammatory myofibroblastic sarcoma (EIMS) has been suggested to distinguish this aggressive variant [43].
In a previous study of 59 IMTs, analysis of a subset of tumours with histological atypia and aggressive clinical course had revealed that ALK negative tumours were associated with older age of presentation, greater nuclear pleomorphism, nuclear atypia and atypical mitosis with a tendency for distant metastasis [44]. In our series, two patients were adult males in the 40–50 age group, while the third was a 16-year-old female. All three had IMTs with IHC positive for ALK protein and FISH detected EML4-ALK1 re-arrangement. While the two older males could be treated with endobronchial resection, the female had a local recurrence. This series is too small to draw firm conclusions about tumour site, molecular profile, histological profile and prognosis.
The molecular genetics of IMT has opened up a potentially new therapeutic possibility in unresectable and refractory tumours. The ALK inhibitor crizotinib has been evaluated in patients with advanced or metastatic IMTs deemed incurable by surgery, radiotherapy or systemic therapy with response rates of 50% by RECIST criteria in ALK-positive and 14% in ALK-negative tumours, concluding that crizotinib should be considered standard of care in the subset of patients with ALK-positive IMTs who do not qualify for curative surgery [7]. However, amongst ALK-negative tumours, the presence of other oncogenic drivers such as ROS1 gene rearrangements may render them amenable to treatment with crizotinib [6]. Ceritinib has been used with a significant and sustained partial response in an IMT that had progressed despite crizotinib [45]. A future perspective may be to study the efficacy of combined modalities including bronchoscopic resection and ALK1 inhibitors, especially in refractory or recurrent tumours.
In conclusion, interventional bronchoscopic techniques are minimally invasive lung function preserving modalities for resection of endobronchial IMTs in adults. They are particularly useful when the tumour has essentially endoluminal localisation and complete resection of the tumour can be performed during the bronchoscopic procedure. Regular follow-up is essential to detect residual tumour or recurrence. It may also prove useful for airway recanalisation in patients who are unfit for surgery or in emergency respiratory distress as a bridge to surgical resection. In unresectable and metastatic cases, targeted treatment with ALK inhibitors may be considered standard of care as guided by the molecular profile of the tumour.
Footnotes
Author contributions: A. Iyer: contributions towards conception, design, drafting and revision of the article and its scientific content; acquisition of clinical data; and final approval of content. T. Radonic: contribution towards design, drafting and revision of the article and its scientific content; acquisition of clinicopathological data; and final approval of content. L.C. Heukamp: contributions towards revision of the article and its scientific content; acquisition of molecular pathological data; and final approval of content. E. Thunnissen: contributions towards design, drafting and revision of the article and its scientific content; acquisition of clinicopathologic data; and final approval of content. J.M.A. Daniels: contributions towards conception, design, drafting and revision of the article and its scientific content; acquisition of clinical data; and final approval of content.
Conflict of interest: A. Iyer has nothing to disclose.
Conflict of interest: T. Radonic has nothing to disclose.
Conflict of interest: L.C. Heukamp has nothing to disclose.
Conflict of interest: E. Thunnissen has nothing to disclose.
Conflict of interest: J.M.A. Daniels has nothing to disclose.
- Received March 27, 2020.
- Accepted November 13, 2020.
- Copyright ©ERS 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










