





Abstract
Background There are no established therapeutic options available for idiopathic pleuroparenchymal fibroelastosis (IPPFE) apart from supportive care and lung transplantation. Furthermore, it is known that IPPFE with a usual interstitial pneumonia (UIP) pattern and lower lobe predominance is a disease entity distinct from idiopathic pulmonary fibrosis (IPF). To our knowledge, few studies are available that report on the efficacy of antifibrotic agents for IPPFE with UIP.
Aim The aim of this study was to compare the efficacy of antifibrotic agents between IPPFE with UIP and typical IPF in real-world clinical practice.
Patients and methods A retrospective analysis was performed on the medical records of all patients at two interstitial lung disease centres. Sixty-four patients were diagnosed as having IPPFE with UIP and 195 patients were diagnosed with typical IPF. We compared the efficacy of antifibrotic agents between these two groups.
Results Survival time was significantly shorter in the patients with IPPFE with UIP. Some 125 patients were administered antifibrotic agents for over 6 months (34 with IPPFE with UIP and 91 with typical IPF). Reduced forced vital capacity (FVC) 6 months after treatment with antifibrotic agents was significantly greater in the IPPFE with UIP group than in those in the typical IPF group. Moreover, the change in % predicted FVC was significantly greater during the follow-up in patients with IPPFE with UIP compared with those with typical IPF.
Conclusions The efficacy of antifibrotic agents was limited in patients with IPPFE with UIP. Thus, IPPFE with UIP remains a fatal and progressive disease.
Abstract
This study demonstrated that patients with IPPFE with UIP have a poorer prognosis than those with typical IPF because these patients were intractable to treatment with antifibrotic agents https://bit.ly/38LbAKD
Introduction
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare condition characterised by predominantly upper lobe pleural and subjacent parenchymal fibrosis [1]. In general, there are no established therapeutic options available for IPPFE other than supportive care and lung transplantation [2].
IPPFE has been increasingly reported in association with a variety of interstitial lung diseases (ILDs) in the lower lobes, including usual interstitial pneumonia (UIP) [3, 4]. Some patients with IPPFE with UIP pattern are diagnosed as having idiopathic pulmonary fibrosis (IPF) and are treated with antifibrotic agents in real-world clinical practice. However, IPPFE with UIP could be a disease entity distinct from IPF because patients with IPPFE with UIP had poorer prognoses compared with those of patients with IPF, as reported by several researchers [5–8].
In recent years, it has been reported that progressive fibrosing ILD other than IPF (non-IPF PF-ILD) is characterised by disease progression associated with worsening of fibrosis despite appropriate treatment for individual patients with ILD [9, 10]. Given patients with non-IPF PF-ILD have shown similarities in underlying pathogenetic mechanisms and disease behaviour to those with IPF, it appears plausible that antifibrotic treatments could be beneficial in these conditions [11, 12]. In fact, Distler et al. [13] had reported the potential efficacy of nintedanib in patients with systemic sclerosis-associated ILD. More recently, the annual rate of decline in forced vital capacity (FVC) was significantly lower among patients with non-IPF PF-ILD who received nintedanib than among those who received placebo in the INBUILD trial [14]. However, there is no evidence for the efficacy of antifibrotic treatment, including nintedanib and pirfenidone, for IPPFE with UIP.
Thus, we aimed to clarify differences in the efficacy of antifibrotic agents and the prognoses between IPPFE with UIP and typical IPF.
Methods
Study population and clinical data
The study population consisted of consecutive patients at the Department of Respiratory Medicine of Tsuboi Hospital and Toho University Omori Medical Centre in Japan from April 2003 to May 2018. The diagnosis of IPPFE with UIP, which was modified from previous studies [3, 15, 16], was determined by the presence of the following features in addition to honeycombing, predominantly in the bilateral lower lobes: 1) bilateral dense subpleural consolidation in the upper lobes with traction bronchiolectasis, architectural distortion and upper lobe volume loss; 2) exclusion of other identifiable aetiologies, such as a history of radiation therapy, active pulmonary infection, connective tissue disease and chronic hypersensitivity pneumonitis; and 3) radiological confirmation of disease progression, defined as an increase in the upper lobe subpleural consolidation and/or a decrease in upper lobe volume on serial chest high-resolution computed tomography (HRCT). We identified IPF in accordance with the 2011 international IPF guidelines [17], and acute exacerbation of IPF was diagnosed by criteria proposed by Collard et al. [18], in which all of the following four conditions must be satisfied: 1) a previous or concurrent diagnosis of IPF; 2) unexplained worsening or development of dyspnoea within 30 days; 3) chest HRCT scan with new bilateral ground-glass opacities and/or consolidation superimposed on a reticular or honeycombing background pattern; and 4) no evidence of pulmonary infection by bronchoalveolar lavage, endotracheal aspiration or sputum culture, in combination with negative blood tests for other potentially infectious pathogens (e.g. Pneumocystis jirovecii, Cytomegalovirus) and exclusion of left heart failure, pulmonary embolism and alternative causes of acute lung injury.
The diagnosis of all patients was evaluated by a multidisciplinary discussion based on patients’ clinical, radiological and/or pathological findings.
The medical records were retrospectively reviewed to obtain the following clinical data: age, sex, smoking status, body mass index (BMI), modified Medical Research Council (mMRC) scale, laboratory data (Krebs von den Lungen 6 (KL-6), surfactant protein D), pulmonary function test findings and chest HRCT findings. We examined the data on the patients’ clinical course, including treatment, complications, changes in pulmonary function (every 6 months) and prognosis. The Gender–Age–Physiology (GAP) score was calculated using the data obtained at the initial evaluation [19]. To evaluate the response to treatment, we defined disease progression as a relative decline in ≥10% and stable disease as a relative decline in <10% in per cent predicted FVC over a period of 6 months.
The Institutional Ethics Committee of the Toho University Omori Medical Centre approved this study (no. M1626317281) and waived the requirement for informed consent given the study was designed as a retrospective clinical review.
Visual computed tomography analysis
Helical computed tomography (CT) scanners (Aquilion 16, Toshiba, Tokyo, Japan; and Aquilion Prime, Canon, Tokyo, Japan) were employed. Thin-section CT scans were obtained at full inspiration, and the scanning protocol consisted of reconstruction of a 1- to 2-mm slice with a high-spatial-frequency algorithm. Thin-section CT images of the chest were photographed at window settings appropriate for the lung parenchyma (window level from –600 to –450 HU; width from 1600 to 1900 HU) for all patients.
Fibrosis score (reticulation and honeycombing) was semi-quantitatively evaluated based on the extent of lung parenchymal involvement in whole lungs: 0 (absent); 1 (<25%); 2 (25% to <50%); 3 (50% to <75%) and 4 (≥75%) [15]. Traction bronchiectasis extent was also scored: 0, none; 1, 1 segment; 2, 2 segments; 3, more than 2 segments [20]. These scores were reviewed independently by two pulmonologists (K.S. and H.O. who had 22 and 8 years of experience in ILD practice, respectively) and one radiologist (K.M. who had 20 years of experience in chest radiology), blinded to the clinical data. The simple kappa value was 0.62 for radiological pleuroparenchymal fibroelastosis (PPFE). The weighted kappa values for fibrosis score and extent of bronchiectasis were 0.85 and 0.87, respectively.
Pulmonary function testing
Spirometry and the diffusing capacity of the lung for carbon monoxide (DLCO) by the single breath-holding method were measured using a pulmonary function test system (Chestac-33 and Chestac-8900; CHEST Co. Ltd., Tokyo, Japan).
Statistical analysis
Data are expressed as means with standard deviations or numbers of patients with percentages, as appropriate. The differences between the two groups were evaluated using Pearson's Chi-squared test or Fisher's exact test for categorical variables, t-test for parametric continuous variables and the Mann–Whitney U-test for nonparametric variables. Mean changes in FVC values for 6 months before and after treatments with antifibrotic agents were compared between the two groups by a two-way repeated measure analysis of variance with Bonferroni's multiple comparison. We compared longitudinal changes in % predicted FVC (%FVC) from baseline by using a linear mixed model (LMM) with random intercept and slope term (R version 3.5.2; R Foundation for Statistical Computing, Vienna, Austria). Fine–Gray univariable and multivariable competing risks models were used to investigate variables predictive of mortality among variables that were demonstrated to be significant in the univariate model. Variables with p<0.1 were used for entry into the model. To avoid multicolinearity, only one of the highly correlated variables (correlation coefficient ≥0.7) was entered into the multivariate model (R version 3.5.2, R Foundation for Statistical Computing). We performed logistic regression analysis to identify predictive factors associated with efficacy of antifibrotic agents. Overall survival was defined as the time from the date of diagnosis to the date of censoring or death. Kaplan–Meier survival curves were compared using the log rank test. We considered p<0.05 to represent statistical significance. We analysed the interobserver variation in various abnormalities on chest HRCT using the kappa (κ) statistic. Interobserver agreement was classified as follows: poor, κ=0–0.20; fair, κ=0.21–0.40; moderate, κ=0.41–0.60; good, κ=0.61–0.80; and excellent, κ=0.81–1.00. All data except for LMM and Fine–Gray univariable and multivariable competing risks models were performed using JMP, version 10.0.0 (SAS Institute Inc., Cary, NC, USA).
Results
Baseline clinical differences between patients with IPPFE with UIP and patients with typical IPF
The proportion of women, the rate of never-smokers and the percentage of pneumothorax complications were significantly higher in patients with IPPFE with UIP than in those with typical IPF. There were no differences in the baseline disease severity (GAP staging) between both groups (stage I/II/III=28/24/12 versus 99/68/28, p=0.55). Conversely, the percentage of primary lung cancer complications was significantly lower in patients with IPPFE with UIP than in those with typical IPF (table 1). Baseline values of %FVC and serum KL-6 values in patients with IPPFE with UIP were significantly lower than those in patients with typical IPF, whereas the level of % predicted residual volume and arterial carbon dioxide tension (PaCO2) were significantly higher in patients with IPPFE with UIP (table 2).
Demographic and baseline patient characteristics
Comparison of pulmonary function tests, serum markers and chest computed tomography (CT) findings between patients with idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP) and typical idiopathic pulmonary fibrosis (IPF)
Overall survival and prognostic significance of patients with IPPFE with UIP
The Kaplan–Meier survival curve had a significantly poorer outcome in patients with IPPFE with UIP (median survival time (MST) 34.0 months versus 62.3 months, p<0.0001) (figure 1). Regarding prognostic factors for survival for patients with IPPFE with UIP, a univariable Fine–Gray competing risks analysis considering acute exacerbation as a competing risk showed that decreased BMI (subdistribution hazard ratio (SHR) 0.757; 95% CI 0.632–0.907; p=0.003), decreased %FVC (SHR 0.955; 95% CI 0.929–0.982; p=0.001) and pneumothorax complication (SHR=3.029; 95% CI 1.366–6.714; p=0.006) were significant predictors. The multivariate Fine–Gray competing risks analysis demonstrated that the prognostic factors were decreased BMI (SHR 0.806; 95% CI 0.659–0.987; p=0.037), decreased %FVC (SHR 0.952; 95% CI 0.920–0.985; p=0.005) and prednisolone use (SHR 0.228; 95% CI 0.072–0.727; p=0.012) (table 3).

The Kaplan–Meier survival curve in patients with typical idiopathic pulmonary fibrosis (IPF) (solid line) (n=195) and idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP) (dashed line) (n=64) (median survival time 62.3 months versus 34.0 months, p<0.0001).
Fine–Gray univariable and multivariable competing risks models demonstrating prognostic factors for survival in patients with idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP) (n=64)
Causes of death
The mortality rates associated with pneumonia and chronic deterioration in IPPFE with UIP were significantly higher than those in typical IPF during follow-up (table 4).
Comparison of causes of death between idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP) and typical idiopathic pulmonary fibrosis (IPF)
Antifibrotic treatments for patients with IPPFE with UIP
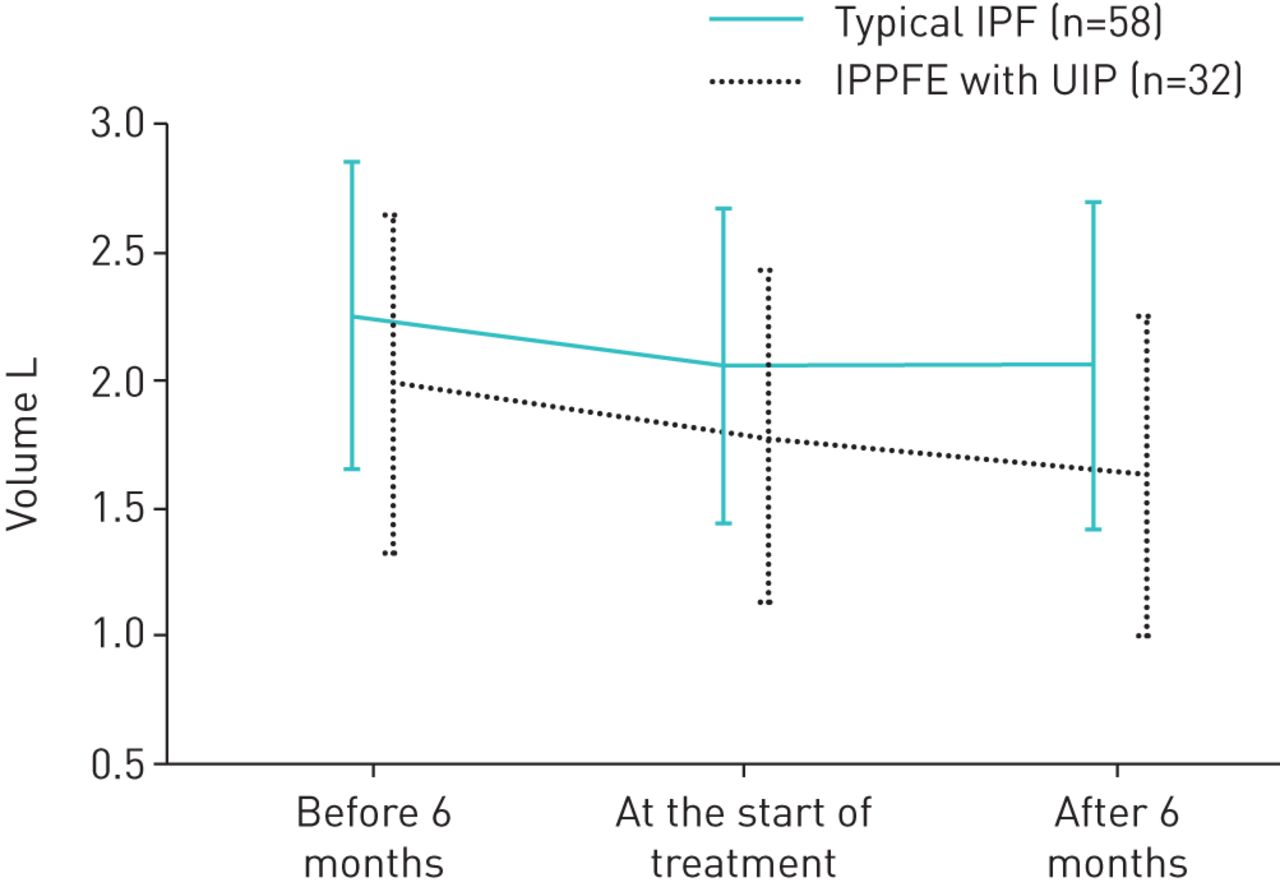
A total of 125 patients were administered antifibrotic drugs for over 6 months (34 with IPPFE with UIP and 91 with typical IPF). Some 26 of 34 patients with IPPFE with UIP and 53 of 91 patients with typical IPF were treated with pirfenidone, and the remaining patients received nintedanib. In terms of baseline clinical differences between patients with IPPFE with UIP and patients with typical IPF who were treated with antifibrotic agents, there were no differences in baseline disease severity (GAP staging) or in radiological scores between the two groups. However, values of %FVC, serum KL-6 and BMI in patients with IPPFE with UIP were significantly lower than those in patients with typical IPF, whereas the level of PaCO2 was significantly higher in patients with IPPFE with UIP. There were no differences between the groups in reduced FVC value 6 months before treatment with antifibrotic agents (IPPFE with UIP versus typical IPF=−0.21±0.16 L versus −0.20±0.19 L; p=0.95). However, more significantly reduced FVC values 6 months after treatment with antifibrotic agents were found in patients with IPPFE with UIP than in those with typical IPF (IPPFE with UIP versus typical IPF=−0.15±0.17 L versus −0.004±0.18 L; p=0.0002) (figure 2). There was no difference in comparative efficacy between pirfenidone and nintedanib in patients with IPPFE with UIP and those with typical IPF (rate of disease progression in IPPFE with UIP: pirfenidone versus nintedanib 35.3% versus 2.9%, respectively; p=0.12; typical IPF: pirfenidone versus nintedanib 5.5% versus 6.6%, respectively; p=0.52). The LMM analysis showed that the decline in the slope of %FVC during follow-up was significantly different between the two groups (p=0.0003) (figure 3a). Moreover, patients with IPPFE with UIP during follow-up who were treated with antifibrotic agents had a significantly lower baseline %FVC and a more rapid decline in FVC compared with that in those with typical IPF treated with antifibrotic agents (p=0.0002) (figure 3b). Additionally, a predictive factor for poor response to antifibrotic agents was the presence of IPPFE with UIP in the multivariate logistic regression analysis (table 5).

Change in forced vital capacity (FVC) before and after treatments with antifibrotic agents (idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP)/typical idiopathic pulmonary fibrosis (IPF)in 32 out of 58 cases). There were no differences between the groups in FVC value 6 months before treatment with antifibrotic agents (IPPFE with UIP versus typical IPF = −0.21±0.16 L versus −0.20±0.19 L; p=0.95). However, more significantly reduced FVC values 6 months after treatment with antifibrotic agents were found in patients with IPPFE with UIP than in those with typical IPF (IPPFE with UIP versus typical IPF = −0.15±0.17 L versus −0.004±0.18 L; p=0.0002; IPPFE with UIP: before 6 months versus at the onset of treatments versus after 6 months = 1.96±0.66 L versus 1.75±0.64 L versus 1.61±0.61 L; p<0.0001, p<0.0001; typical IPF: before 6 months versus at the onset of treatments versus after 6 months = 2.25±0.61 L versus 2.05±0.62 L versus 2.05±0.64 L, p<0.0001, p=1.00). Two-way repeated measure ANOVA with Bonferroni's multiple comparison.

{kind=link}
{kind=link}
{kind=link}
Change in % predicted forced vital capacity (FVC) during follow-up. a) Linear mixed model (LMM) analysis showed that the decline in the slope of FVC during follow-up was significantly different between the two groups (p=0.0003). Patients with typical idiopathic pulmonary fibrosis (IPF) had a significantly higher baseline FVC than those with idiopathic pleuroparenchymal fibroelastosis (IPPFE) with usual interstitial pneumonia (UIP) (77.4±18.7% versus 63.9±15.7%, p<0.0001). b) Patients with IPPFE with UIP during follow-up who were treated with antifibrotic agents had a significantly lower baseline FVC and a more rapid decline in FVC compared with that in those with typical IPF treated with antifibrotic agents (p=0.0002). Patients with typical IPF had a significantly higher baseline FVC than those with IPPFE with UIP (73.8±17.8% versus 64.6±14.9%, p=0.003).
Multivariate logistic regression analysis of predictive factors for efficacy of antifibrotic agents (n=125)
Discussion
This is the first study to compare the efficacy of antifibrotic agents such as pirfenidone and nintedanib between IPPFE with UIP and typical IPF in real-world clinical practice.
IPPFE is a rare condition characterised by predominantly upper lobe pleural and subjacent parenchymal fibrosis according to the 2013 American Thoracic Society/European Respiratory Society classification of idiopathic interstitial pneumonias [1]. These distinctive conditions had first been reported as Amitani's disease or idiopathic pulmonary upper lobe fibrosis (IPUF) by Amitani et al. in 1992 [21], as well as initial case series in the English literature by Frankel et al. [22], which was described as an idiopathic pleuroparenchymal fibroelastotic syndrome with unique histological findings. The prognosis of IPPFE tends to be poor. Watanabe et al. [23] had reported that patients with IPUF were shown to have a histopathological tendency for a relatively rapid progression and a poor prognosis, with a survival time of 1.8 to 12.2 years after their initial visits. IPPFE has been increasingly reported in association with a variety of ILDs in the lower lobes, including UIP [3, 4]. Nakatani et al. [4] reported 12 cases out of 205 patients with ILD who were identified as having IPPFE, 11 of whom had ILD in the lower lobes (definite UIP, 5; possible UIP, 4; nonspecific interstitial pneumonia, 1; unclassifiable, 1).
Histopathologically, biopsied lung specimens can show mild changes in PPFE or other patterns such as UIP in patients with PPFE [1]. Raghu et al. [24] reported that some patients with IPPFE with a UIP pattern should be regarded as having IPF after a multidisciplinary discussion based on the 2018 IPF guidelines. However, considering that the clinical characteristics such as the proportion of women, never-smokers, lower BMI and pneumothorax complications were significantly higher in patients with IPPFE with UIP than in those with typical IPF in the present study, IPPFE with UIP may be treated as a disease entity distinct from typical IPF. As reported by several researchers, patients with IPPFE with UIP had a poor prognosis compared with those with IPF [6–8]. Recently, Oda et al. [6] described how survival time tended to be shorter in patients with PPFE with UIP (MST 31.5 months versus 82.1 months). More recently, Kato et al. [7] reported that survival time for patients with IPPFE with UIP was significantly shorter than that for patients without UIP (MST 12 months versus 62 months). We have already reported that compared with patients with typical IPF, poorer survival was noted in those with atypical IPF who had imaging features on chest HRCT, such as PPFE-like lesions or multiple thick-walled large cysts, in addition to honeycombing in the bilateral lower lobe [20]. Additionally, in the present study, patients with IPPFE with UIP have a poorer prognosis than those with typical IPF (MST 34.0 months versus 62.3 months). In patients with IPPFE with UIP, decreased BMI and %FVC were associated with a significantly worse survival on both univariable and multivariable Fine–Gray competing risks analysis that considered acute exacerbation as a competing risk. These results support the argument that survival in patients with IPPFE with UIP is associated with chronic deterioration. Moreover, a significantly faster decline in FVC was observed in patients with IPPFE with UIP after antifibrotic treatments, whereas the FVC decline was suppressed in patients with typical IPF after 6 months of receiving antifibrotic agents. Notably, LMM analysis demonstrated that a decline was significantly steeper in %FVC during a follow-up in patients with IPPFE with UIP treated with antifibrotic agents compared with those with typical IPF. Although pirfenidone was described by Sato et al. [25] as potentially effective in a case report, our data suggest that the efficacy of antifibrotic treatments might be limited for IPPFE with UIP. Also, we suppose that the higher incidence of pneumothorax complications in IPPFE with UIP can effect a faster FVC decline in addition to being intractable to antifibrotic treatments.
Distler et al. [13] reported the potential efficacy of nintedanib in patients with systemic sclerosis-associated ILD. More recently, the annual rate of decline in FVC was significantly lower among patients with non-IPF PF-ILD who received nintedanib than among those who received placebo in the INBUILD trial [14]. However, there is no evidence suggesting efficacy of antifibrotic treatment, including nintedanib and pirfenidone, for IPPFE with UIP. In our study, the multivariate logistic regression analysis showed that the presence of IPPFE with UIP was a predictive factor for poor response to antifibrotic agents.
There are some limitations to our study. First, this was a retrospective study with a relatively small sample size. Therefore, our results might not be representative of the entire IPPFE with UIP population and should be confirmed in a larger cohort. Second, most of the patients with IPPFE with UIP could not be diagnosed histologically, as surgical lung biopsy can trigger a pneumothorax [26]. Furthermore, patients with a clinical diagnosis of PPFE had similar characteristics to those of histopathologically confirmed PPFE, as described by Enomoto et al. [27]. Therefore, we diagnosed IPPFE with UIP primarily according to radiological criteria in this study. Third, it is difficult to differentiate between PPFE lesions and apical cap on chest HRCT. However, we believe that the presence of traction bronchiectasis and volume loss in the upper lobes is useful in differentiating these conditions. Finally, this study did not investigate the comparative efficacy of nintedanib and pirfenidone. Further large-scale studies are needed to explore this issue.
In conclusion, this study demonstrated that the efficacy of antifibrotic agents was limited in patients with IPPFE with UIP. Thus, IPPFE with UIP remains a fatal and progressive disease. Future studies are required including prospective analyses of the efficacy of antifibrotic agents for patients with IPPFE with UIP.
Acknowledgements
We would like to thank Chiaki Nishimura, Professor Emeritus of Toho University and Representative of CN Medical Research for helpful discussions and statistical analysis.
Footnotes
Author contributions: Study design and concept: K. Sugino, H. Ono, S. Homma and K. Kishi; patient data collection: K. Sugino, H. Onon, H. Shimizu, T. Kurosawa, K. Matsumoto, M. Ando and K. Mori; data analysis: K. Sugino, H. Ono and K. Kishi; manuscript preparation and review: K. Sugino, H. Ono, T. Kurosawa, E. Tsuboi, S. Homma and K. Kishi.
Conflict of interest: K. Sugino reports a lecture fee from Nippon Boehringer Ingelheim Co., Ltd., outside the submitted work.
Conflict of interest: H. Ono has nothing to disclose.
Conflict of interest: H. Shimizu has nothing to disclose.
Conflict of interest: T. Kurosawa has nothing to disclose.
Conflict of interest: K. Matsumoto has nothing to disclose.
Conflict of interest: M. Ando has nothing to disclose.
Conflict of interest: K. Mori has nothing to disclose.
Conflict of interest: E. Tsuboi has nothing to disclose.
Conflict of interest: S. Homma reports a lecture fee from Nippon Boehringer Ingelheim Co., Ltd., outside the submitted work; and is a member of an endowed department sponsored by Teijin Pharma, Co., Ltd, Nippon Boehringer Ingelheim, Co., Ltd, Shionogi & Co., Ltd, Chugai Pharmaceutical Co., Ltd, and Asahi Kasei Pharma Co., Ltd.
Conflict of interest: K. Kishi reports a lecture fee from Nippon Boehringer Ingelheim Co., Ltd., outside the submitted work.
Support statement: This work was supported by a grant-in-aid for diffuse lung diseases from the Japanese Ministry of Health, Labor and Welfare.
- Received April 19, 2020.
- Accepted November 5, 2020.
- Copyright ©ERS 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










