





Abstract
Background COPD patients often have cardiac comorbidities. Cardiac involvement at the time of a COPD exacerbation is associated with a high short-term mortality, but whether this influences long-term outcomes is unknown. We explored whether biomarkers of cardiac dysfunction at the time of a COPD exacerbation predict long-term outcomes.
Methods Two prospective cohorts of patients admitted to Waikato Hospital for exacerbations of COPD were recruited during 2006–2007 and 2012–2013. N-terminal pro-B-type natriuretic peptide (NT-proBNP) and troponin T were measured on admission and were used to indicate cardiac stretch and myocardial injury, respectively. 5-year survival after discharge and subsequent admissions for cardiac disease and COPD exacerbations were analysed using Kaplan–Meier and Cox proportional hazards tests.
Results The overall 5-year mortality was 61%. Patients with high NT-proBNP on admission had higher mortality than those with normal cardiac biomarkers (adjusted hazard ratio (aHR) 1.76, 95% CI 1.18–2.62). High NT-proBNP was also associated with a higher risk of future cardiac admissions (aHR 1.75, 95% CI 1.2–2.55). Troponin T levels were not associated with long-term survival (aHR 0.86, 95% CI 0.40–1.83) or future cardiac admissions (aHR 0.74, 95% CI 0.34–1.57). Neither biomarker predicted future COPD exacerbations.
Conclusion The long-term prognosis following a hospitalisation for an exacerbation of COPD is poor with less than half of patients surviving for 5 years. Elevated NT-proBNP at the time of a COPD exacerbation is associated with higher long-term mortality and a greater likelihood of future cardiac admissions, but not future COPD exacerbations.
Abstract
Elevated NT-proBNP at the time of #AECOPD is associated with higher long-term mortality and a greater likelihood of future cardiac admissions, but not future COPD exacerbations https://bit.ly/37wnRQP
Introduction
Cardiovascular disease is an important comorbidity in COPD patients and is estimated to cause up to 37% of deaths [1, 2]. People with COPD have a high risk of cardiovascular disease because of the shared risk factors of advancing age and smoking, which contribute to the pathophysiology of both conditions [3]. However, lung and cardiovascular disease frequently coexist even in nonsmokers, indicating that there are other mechanisms linking these diseases.
Cardiac comorbidities are particularly important in the setting of acute exacerbations of COPD. Several studies have documented high rates of myocardial infarction, arrhythmias and heart failure among patients experiencing exacerbations of COPD, and these are associated with a worse short-term prognosis [4–9]. Exacerbations of COPD, heart failure and acute coronary syndromes can all present as dyspnoea or chest discomfort, and it is likely that cardiovascular disease is underdiagnosed in patients with COPD exacerbations leading to undertreatment [4].
We have previously reported on two prospective observational cohort studies of biochemical evidence of cardiac pathology in patients admitted to hospital for acute exacerbations of COPD without clinical evidence of acute cardiac disease [10–13]. The first study (cohort 1) found that 28% of participants had elevated N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels and 17% had elevated troponin T [10]. Elevated cardiac biomarkers were associated with substantially higher 30-day mortality. These findings were confirmed in the second study (cohort 2) [12]. These biomarkers appear to indicate acute or chronic cardiac involvement that cannot be reliably detected by clinical assessment, routine chest radiographs or electrocardiograms [13]. Furthermore, the elevated cardiac biomarkers are associated with both left and right heart dysfunction [14]. Thus, cardiac involvement has at least short-term prognostic significance in the setting of COPD exacerbations.
The long-term prognosis after hospitalisation for an exacerbation of COPD is poor, with a 5-year mortality of around 60% [4]. It is unknown to what extent subclinical cardiac involvement influences this prognosis beyond the acute phase of the COPD exacerbation.
To test the hypothesis that elevated biomarkers of cardiac injury (troponin) and cardiac stretch (NT-proBNP) during exacerbations of COPD would predict long-term prognosis for survival, further COPD exacerbations and future cardiac events, we combined and analysed the data from these two prospective cohorts.
Methods
Study participants
Two cohorts of consecutive patients admitted to Waikato Hospital for exacerbations of COPD were recruited (cohort 1, n=247 (2006–2007) and cohort 2, n=176 (2012–2013)). The details of these cohorts have been published previously [10, 11]. Each cohort recruited patients over 1 complete year. Patients who did not live locally or could not attend follow-up were excluded. Both cohorts recruited all consenting patients with clinically diagnosed exacerbations of COPD but excluding those with suspected acute cardiac disease and those with pneumonia (consolidation on chest radiographs). The second cohort also excluded patients with end-stage renal disease. For those with multiple admissions over the year, the first admission was regarded as the index admission.
All participants gave written consent at the time of study inclusion, and approval was obtained for both cohorts from the New Zealand Health and Disability Ethics Committee.
Data collection
For this analysis the electronic records of patients were accessed to ascertain mortality within 5 years of discharge from hospital. We also recorded readmissions for exacerbations of COPD and cardiac admissions with heart failure or acute coronary syndromes within the Waikato District Health Board region. Fourteen participants recruited in both cohort 1 and 2 were removed from cohort 2 to avoid duplication.
Cardiac biomarkers
Both cohorts collected blood for measurement of NT-proBNP and cardiac troponin T within 24 h of admission to hospital. Measurements of NT-proBNP and troponin T were performed by Waikato Hospital laboratory using quantitative electrochemiluminescence assay. NT-proBNP levels in both cohorts had a detection limit of 0.6 pmol·L−1 (Electsys proBNP, Roche Diagnostics Corporation). The troponin detection limit was 0.01 µg·L−1 for cohort 1 and 5 ng·L−1 for cohort 2, because a high sensitivity troponin assay was introduced between the cohorts (troponin and TnT-hs, Roche Diagnostics Corporation).
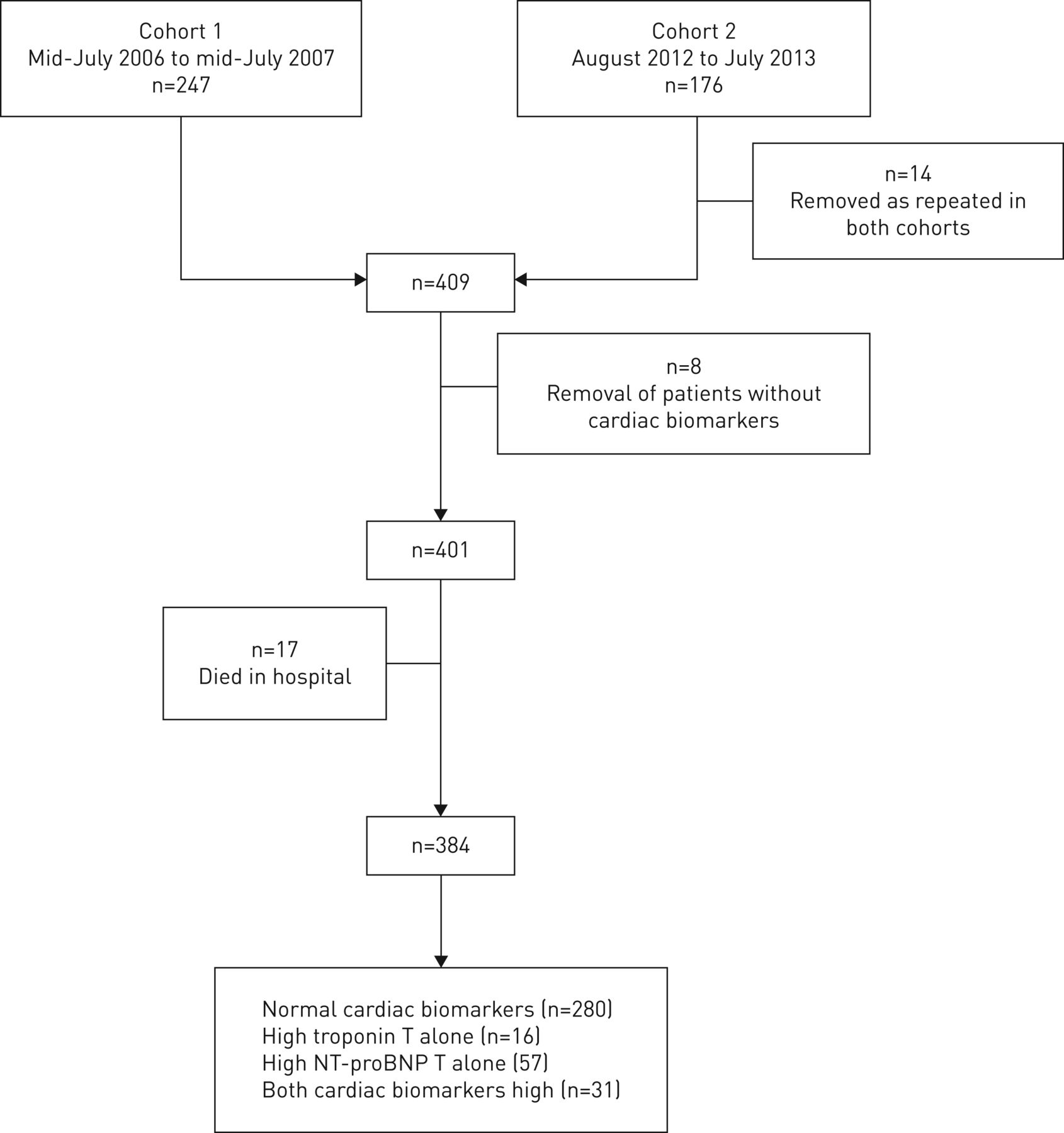
NT-proBNP >220 pmol·L−1 and troponin T >0.03 μg·L−1 in cohort 1 and >50 ng·L−1 in cohort 2 are considered abnormal according to local laboratory reference values. These levels were used to group participants into normal cardiac biomarkers, high troponin T only, high NT-proBNP only and both high troponin T and NT-proBNP groups (figure 1).
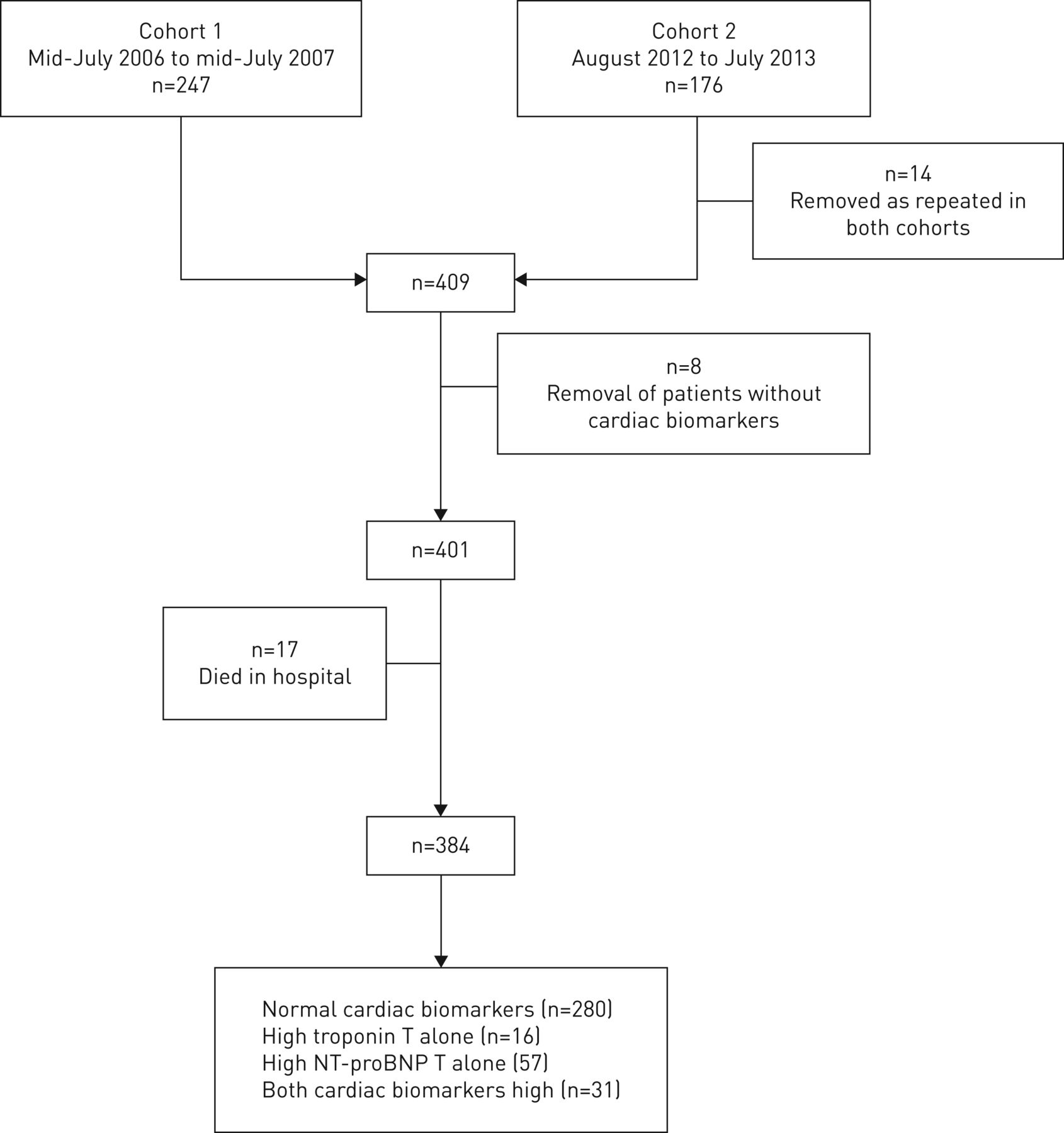
Study design. Patients who died in hospital (n=17) were included in overall mortality data but were excluded from survival analyses after discharge. NT-proBNP: N-terminal pro-B-type natriuretic peptide.
Statistical analysis
The primary end point was survival after discharge. Patients who died during their index admission were excluded from the analyses. This was plotted using Kaplan–Meier survival curves for the groups with normal or abnormal biomarkers. Cox proportional hazard ratios (HRs) were calculated with and without adjustment for acute COPD severity using the CURB-65 (Confusion, Urea, Respiratory rate, Blood pressure, Age ≥65) score and acidaemia (blood pH <7.30) on admission, and severity of airflow obstruction using forced expiratory volume in 1 s (FEV1 % pred) [10, 12]. Secondary end-points were readmissions for exacerbations of COPD or acute cardiac disease. Because patients may have died before readmission, these were calculated as COPD admission-free survival and cardiac admission-free survival, respectively, with follow-up censored at the date of death. Because these analyses used pre-existing cohorts, no formal sample size calculations were undertaken. Analyses were conducted with Stata (v.16; StataCorp, College Station, TX, USA).
Results
The contribution of each cohort is shown in figure 1. The demographic and clinical characteristics of the participants are shown in table 1. Detailed baseline characteristics have been published previously [10, 11] and are available in tables S1 and S2. Including those who died during the index admission, 7%, 33% and 61% of patients died in the first 30 days, 2 years and 5 years, respectively (figure 2). Risk factors for death at 5 years after index admission are shown in table S3 and these include a history of cardiac disease recorded at the time of the index admission.
Baseline characteristics
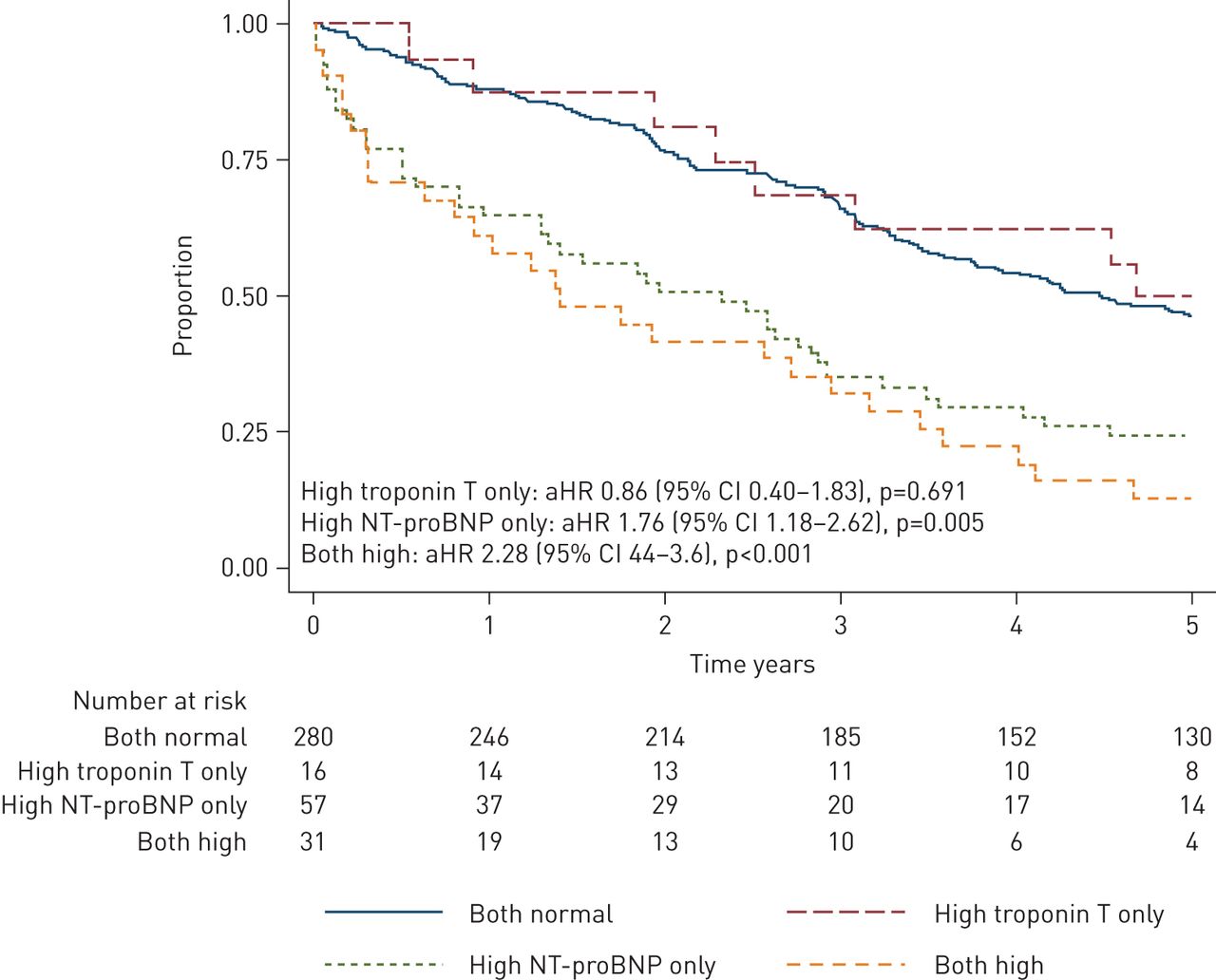
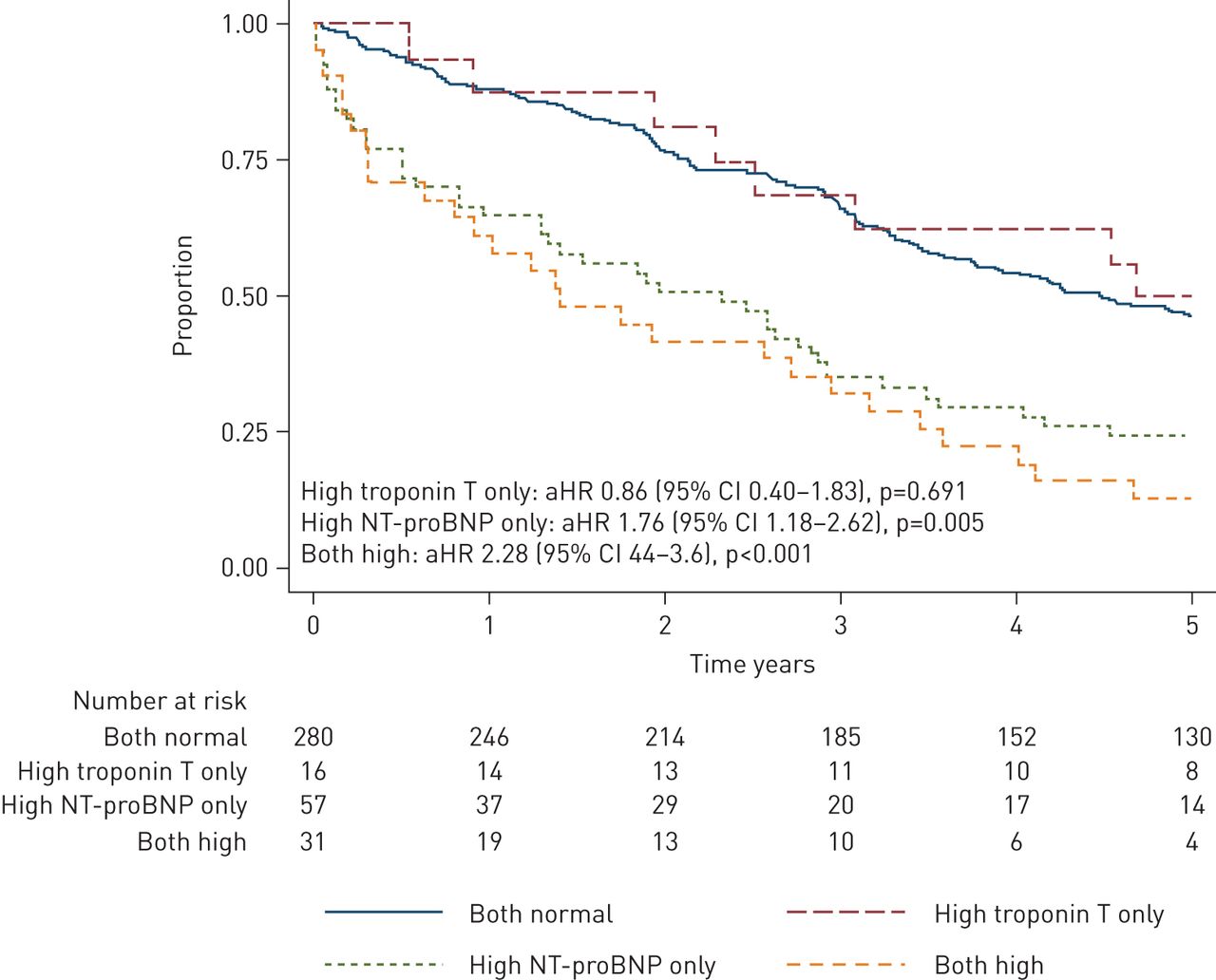
Kaplan–Meier curve of survival over 5 years after discharge for exacerbation of COPD. HR adjusted for CURB-65, acidaemia (pH<7.30) and forced expiratory volume in 1 s (% pred). aHR: adjusted hazard ratio; NT-proBNP: N-terminal pro-B-type natriuretic peptide.
Kaplan–Meier survival curves for those who survived to discharge from the index admission (n=384) for each cardiac biomarker group are shown in (figure 2). Visual inspection indicated that the proportional hazards assumption was met, and the Cox proportional HRs are shown in table 2. Compared to participants who had normal cardiac biomarker measurements, participants with elevated NT-proBNP only had 2.13 times higher mortality during this period. This remained the case after adjusting for exacerbation severity (acidaemia and CURB-65) and baseline lung function (FEV1 % pred) (table 2). By contrast elevated troponin T only in participants was not associated with a worse 5-year survival compared to those with normal biomarkers, and participants with abnormalities of both NT-proBNP and troponin T had similar survival to those who only had abnormal NT-proBNP (figure 2, table 2). These findings were similar after adjusting for a history of cardiac disease (table S4).
Combined cohort adjusted and unadjusted hazard ratios (HRs) for 5-year survival, exacerbation-free survival and cardiac admission-free survival following discharge after index COPD admission according to cardiac biomarker status
Cardiac biomarkers were not associated with future COPD exacerbations (figure 3). However, compared to participants who had normal cardiac biomarker measurements, participants with elevated NT-proBNP alone or both elevated NT-proBNP and troponin T were more likely to be admitted for cardiac disease within 5 years (figure 4). Elevated troponin T alone was not associated with future cardiac admissions.

Kaplan–Meier curve of subsequent COPD hospitalisations following discharge after index COPD exacerbation. HR adjusted for CURB-65, acidaemia (pH<7.30) and forced expiratory volume in 1 s (% pred). aHR: adjusted hazard ratio; NT-proBNP: N-terminal pro-B-type natriuretic peptide.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curve of subsequent cardiac hospitalisations over 5 years following discharge after index COPD exacerbation. HR adjusted for CURB-65, acidaemia (pH<7.30) and forced expiratory volume in 1 s (% pred). aHR: adjusted hazard ratio; NT-proBNP: N-terminal pro-B-type natriuretic peptide.
Discussion
We have found that an elevated NT-proBNP is a strong predictor of poor long-term prognosis among patients admitted to hospital with an exacerbation of COPD. The overall mortality rate was nearly twice as high among those with elevated NT-proBNP even after adjusting for lung function and exacerbation severity (figure 2, table 2). Elevated troponin T, however, did not predict long-term mortality. These findings suggest that subclinical cardiac dysfunction, but not necessarily acute cardiac injury, is an important determinant of long-term mortality in the setting of COPD exacerbations.
We have previously reported that elevated levels of NT-proBNP and troponin T and their combination were associated with a high short-term (30-day) mortality in cohort 1 [10]. In that analysis we did not find that either biomarker was associated with subsequent survival between 30 days and 1 year. This new analysis extends these findings by combining data from two prospective cohorts with much longer follow-up. The findings show that elevated NT-proBNP is strongly associated with both short- and long-term prognosis after an exacerbation of COPD. NT-proBNP is released by the myocardium as a result of myocardial stretch and is a sensitive indicator of cardiac failure [15]. We also found that high levels of NT-proBNP predicted future admissions for cardiac disease but not future COPD admissions (figures 3 and 4). Hence, subclinical evidence of cardiac dysfunction at the time of an exacerbation may indicate poor cardiac reserve and be a harbinger of future cardiac events. Cardiac dysfunction did not predict acute COPD exacerbations suggesting that poor cardiac reserve may not be an aetiological factor in COPD exacerbations.
In the previous analyses of these cohorts [10, 12], a history of cardiac disease was not a predictor of 30-day or 1-year mortality. However, an analysis of this combined cohort shows that a history of cardiac disease and treatment with cardiac medications (notably diuretics) were associated with higher long-term mortality at 5 years (table S3). The associations of the biomarkers with 5-year survival remained similar after adjustment for a history of cardiac disease, suggesting that acute cardiac dysfunction during exacerbations of COPD is associated with a poor long-term prognosis independently of a known cardiac disease.
While troponin T levels on admission were associated with short-term mortality in these cohorts and have been linked to worse prognosis in other studies, they did not predict long-term mortality, nor did they predict future cardiac admissions in these cohorts [4, 16]. Cardiac troponins form part of the diagnosis of myocardial infarction but are sensitive markers of myocardial injury of any cause, including acute compromise of the pulmonary circulation [15–17]. Our observation that troponin is associated with short-term but not long-term mortality is probably best explained by troponins being released as a consequence of the acute COPD exacerbation causing transient cardiac myocyte injury, but this does not necessarily reflect clinically important underlying cardiac disease. There was no evidence that the raised levels of troponin T were caused by acute coronary syndromes in the study: those with clinically suspected acute coronary syndromes were excluded, and there was little electrocardiographic evidence of cardiac ischaemia. Having a raised troponin level does not necessarily mean an acute coronary syndrome, even among those with a known history of coronary artery disease, as the diagnosis requires a compatible clinical history, serial changes of troponin and dynamic electrocardiographic changes. In most patients with raised troponins there is insufficient evidence to support the diagnosis of acute coronary syndrome [18], and we have previously shown that ECG and chest radiographs often do not detect subclinical biomarker derangements [13]. An additional analysis of these cohorts also revealed that a history of cardiac disease was not associated with a high troponin level on admission (OR 1.22, 95% CI 0.69–2.14, p=0.499).
There is no universally accepted biological mechanism linking COPD and heart disease although there are several plausible explanations. COPD and cardiac disease share many risk factors such as smoking, older age and a chronic inflammatory state [19]. COPD is associated with atherosclerosis and arterial stiffness [20, 21], which increase the risk of cardiac ischaemia [22]. COPD exacerbations are associated with elevated systemic inflammatory markers such as C-reactive protein, interleukin-6 and platelet activation, which are also linked to atherosclerosis [23–26]. Our findings suggest that while an acute insult (i.e. the index admission for COPD) may lead to a rise in troponin levels, only those with cardiac remodelling and reduced cardiac reserve indicated by a high NT-proBNP have a worse long-term prognosis.
Exacerbations of COPD are known to increase systemic sympathetic tone leading to higher metabolic demands of tissues and increasing the risk of cardiac ischaemia [27]. Furthermore, severe exacerbations of COPD are usually treated with high doses of β2 agonists, which stimulate cardiac β2 receptors leading to increases in heart rate [28] and increasing cardiac stress.
Lung hyper-inflation caused by COPD is associated with reduced cardiac exercise tolerance so could contribute to cardiac comorbidity in COPD patients [29]. Pulmonary hypertension is estimated to affect around 30% of patients with moderate and severe COPD [30], causing right ventricular dilatation and hypertrophy, leading to restriction of the left ventricle resulting in a lower cardiac output [31].
Many of these inflammatory and physiological mechanisms are likely to worsen during exacerbations of COPD. We have previously found that NT-proBNP continues to rise during the first few days of a COPD exacerbation but falls as the patient recovers a few weeks later [11]. Whether NT-proBNP during stable COPD is an indicator of long-term prognosis is not yet known [32]. As up to half of COPD patients have left ventricular dysfunction, further investigations into this biochemical marker of cardiac overload may provide valuable insights into the complex interplay of heart–lung pathophysiology [33–35].
A major strength of this study is that these are large, unselected, prospectively recruited cohorts of patients with exacerbations of COPD, with near-complete follow-up data. The outcomes were objective (mortality or readmission). We were also able to adjust for confounders by the severity of the exacerbation and lung function impairment in the analyses. A limitation is that both cohorts are from a single centre; however, this reduces the potential for confounding. The overall 5-year mortality of 61% is similar to other cohorts [4], suggesting that our findings are likely to be generalisable to other centres.
There were some differences between the cohorts: cohort 2 was smaller and excluded patients with severe renal disease or terminal malignancy meaning that patients recruited into cohort 2 were healthier than cohort 1. Participants in cohort 2 were less likely to die in 5 years (HR 0.71, 95% CI 0.54–0.93, p=0.013), but there was no difference between cohorts once adjusted for CURB-65, pH and FEV1 % pred (adjusted HR 0.95, 95% CI 0.70–1.29, p=0.75). The findings were similar for each cohort if analysed separately, hence it seems reasonable to combine the cohorts for the ease of interpretation. In both cohorts, the patients were treated according to best practice at time of admission, and participation in these studies did not influence treatment. The absolute number of patients with elevated biomarkers is also low, which might influence the results of analyses in particular those for troponins. Other limitations of this study include the possibility that some readmissions were not added to the electronic database if these occurred outside the Waikato region, although all deaths are recorded regardless of where they occurred. Another limitation is that although cardiac medications on admission were recorded, data on subsequent cardiac medications received during the follow-up period were not. Therefore, we were unable to analyse the impact of cardiac medications on long-term outcomes following an exacerbation of COPD. The frequency of COPD exacerbations before index admission was also not recorded and therefore was not accounted for in the analyses. A further limitation is that the treating clinicians were not blinded to the results of the cardiac biomarkers and that this information may have influenced their diagnosis of acute cardiac disease and management. This might be expected to reduce the association between biomarkers and mortality, but it is possible that knowledge of a previously abnormal NT-proBNP influenced clinicians to diagnose future events as being of cardiac origin, inflating the association between NT-proBNP and cardiac admissions. The exact causes of death were also not available, and we were unable to analyse whether they differed between biomarker groups.
These findings highlight the critical importance of cardiac comorbidity in COPD. This could be even more important at the time of a COPD exacerbation but may not be recognised. Clinicians should be aware of this and carefully look for cardiac involvement. Whether early diagnosis and treatment of cardiac disease in patients with COPD could improve both long- and short-term outcome is unproven, but identifying subclinical cardiac disease will likely help the assessment of prognosis. Future research is needed to better characterise the relationship and understand the mechanisms of cardiac dysfunction in COPD. It would be of particular interest to determine whether cardiac dysfunction is the cause of higher mortality in COPD or whether it is simply a marker of more severe COPD. Our finding that cardiac dysfunction during a COPD exacerbation is associated with future cardiac admissions but not future COPD admissions signals that treatments targeting cardiac dysfunction may improve outcomes in this population by reducing cardiac rather than respiratory events.
In summary, exacerbations of COPD have a poor long-term prognosis with more than half of patients dying within 5 years of hospitalisation. Abnormal levels of NT-proBNP, a marker of cardiac stretch and cardiac failure, at the time of the COPD admission are associated with higher long-term mortality and subsequent admissions for cardiac disease.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00531-2020.SUPPLEMENT
Acknowledgements
We would like to thank the study participants. We also thank Manisha Cooray, summer student for data collection.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Data availability: individual deidentified participant data will only be available if requested.
Support statement: This study was funded by a summer-studentship grant from the Waikato Medical Research Foundation. Initial recruitment and data collection for cohorts 1 (COPDEC) and 2 (BREATHE) were funded by the Waikato Respiratory Research Fund and the Waikato Medical Research Foundation. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: E. Shafuddin has nothing to disclose.
Conflict of interest: S.M. Fairweather has nothing to disclose.
Conflict of interest: C.L. Chang has nothing to disclose.
Conflict of interest: C. Tuffery has nothing to disclose.
Conflict of interest: R.J. Hancox has nothing to disclose.
- Received July 28, 2020.
- Accepted November 26, 2020.
- Copyright ©ERS 2021
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










