





Abstract
Chronic breathlessness, persistent and disabling despite optimal treatment of underlying causes, is a prevalent and frightening symptom and is associated with many emergency presentations and admission to hospital. Breathlessness management techniques used by paramedics may reduce the need for conveyance to hospital. The Breathlessness RElief AT HomE study (BREATHE) aims to explore the feasibility of conducting a definitive cluster randomised controlled trial (cRCT) for people with acute-on-chronic breathlessness who have called an ambulance, to evaluate the effectiveness and cost-effectiveness of a paramedic-administered non-pharmacological breathlessness intervention.
The trial is a mixed-methods feasibility cRCT. Eight paramedics will be randomised 1:1 to deliver either the BREATHE intervention in addition to usual care or usual care alone at call-outs for acute-on-chronic breathlessness. Sixty participants will be recruited to provide access to routine data relating to the index call-out with optional follow-up questionnaires at 14 days, 1 month and 6 months. An in-depth interview will be conducted with a subgroup. Feasibility outcomes relating to recruitment, data quality (especially candidate primary outcomes), and intervention acceptability and fidelity will be collected as well as providing data to estimate a sample size for a definitive trial.
Yorkshire and The Humber–Sheffield Research Ethics Committee approved the trial protocol (19/YH/0314). The study results will inform progression to, or not, and design of a main trial according to predetermined stop-go criteria. Findings will be disseminated to relevant stakeholders and submitted for publication in a peer-reviewed journal.
Abstract
Acute-on-chronic breathlessness initiates many emergency presentations. The BREATHE protocol describes a feasibility, cluster randomised controlled trial of a paramedic breathlessness management intervention. https://bit.ly/2LZg72w
Introduction
Chronic breathlessness, persistent and disabling despite treatment of underlying causes [1], is a prevalent and often frightening feature in cardiorespiratory disease(s). It is more common in older adults [2] with widespread impacts for patients, family carers and health systems [1–3]. Acute worsening of chronic breathlessness (acute-on-chronic breathlessness) is mostly triggered by physical and/or emotional exertion [4]. Tailored non-pharmacological interventions are effective [5] and include breathing retraining, relaxation and anxiety management techniques, pacing and prioritisation [6], and cool facial airflow, for example, from a hand-held, battery-operated fan [7].
Severe episodes of acute-on-chronic breathlessness may be caused by a worsening/complication of the underlying disease, or when distress perpetuates and magnifies the symptom [8]. Acute-on-chronic breathlessness often triggers emergency use of health services [9]. However, approximately a third of those attending the emergency department (ED) because of acute-on-chronic breathlessness do not need hospital admission, and some ED attendances might be preventable [9]. Estimates of breathlessness as a primary reason for adult ED presentations range between 2.7% and 9% [10–13]. In one UK study, acute-on-chronic breathlessness was a reason for ED conveyance by ambulance in 20% of attendances [9]. The presence and intensity of breathlessness on ED arrival predicts hospital admission [14] and return presentations [15].
For many, the ED is necessary. For others, particularly those with advanced disease where palliation is the priority, the ED is less likely to be the optimal place for care if community-based care is working effectively [16]. People with recurrent acute-on-chronic breathlessness may have anxiety playing a significant role, and for whom targeted community-based management plans may reduce the need for ED attendances [17].
The American Thoracic Society (ATS) recommends a dual approach to breathlessness management [18]. Initial management should be given by first responders, using evidence-based non-pharmacological breathlessness interventions. In addition, patients and carers should receive education and training in appropriate self-management techniques to reduce the need for external help. These techniques should be reinforced at every breathlessness encounter [18]. For some, an acute worsening of breathlessness can become a “teachable moment” [19]. Carers may also learn techniques by observing skilled paramedics [20] in this teachable moment. With this approach, more people with acute-on-chronic breathlessness might be managed safely in the community or, if hospital admission is needed, have their breathlessness brought under control more quickly.
Study aim and objectives
The Breathlessness RElief AT HomE study (BREATHE) aims to define the feasibility and desirability of conducting a definitive cluster randomised controlled trial (cRCT) for people with acute-on-chronic breathlessness to evaluate the effectiveness and cost-effectiveness of a paramedic-administered, non-pharmacological breathlessness intervention.
Recruitment and retention rates
The feasibility of recruiting the required number of paramedic-participants and patient-participants needed for a definitive trial within a reasonable trial timeline will be assessed. This will include the acceptability to be randomised within the paramedic-participants and the feasibility of consenting the patient-participants within the time constraints of clinical priorities during an acute call-out.
Intervention
The acceptability, adherence and fidelity, and safety of the BREATHE intervention will be assessed.
Data quality
The quality of data collected during the paramedic call-out in terms of amount and pattern of missing data will be assessed.
Outcomes
The most clinically relevant primary outcome for the definitive trial will be determined by assessing data completion of candidate primary outcomes, qualitative interview data relating to patient views on relevance and acceptability, and the variability around baseline measures. Using the variability values for primary outcome finally chosen, a sample size estimation for a definitive trial will be made.
Implementation issues
Any issues that could impact the implementation of the intervention will be identified, and this information will be used to inform the development of the definitive trial. This will include the development of training materials for paramedics and subsequent implementation into clinical practice.
Methods and analysis
Design
BREATHE is a mixed-methods feasibility cRCT with an embedded normalisation process theory-based (NPT) study. Paramedics, who will act as the cluster unit, will be randomised 1:1 to deliver either the BREATHE intervention plus usual care or usual care only during call-out to patients experiencing acute-on-chronic breathlessness.
This feasibility trial is community-based, with paramedics recruited from ambulance stations within one regional ambulance service. The first paramedic-participants were randomised in January 2020, with the first patient-participant recruited in February 2020.
The trial procedures for patient-participants are outlined in the study flowchart (figure 1) and schedule of events (table 1).
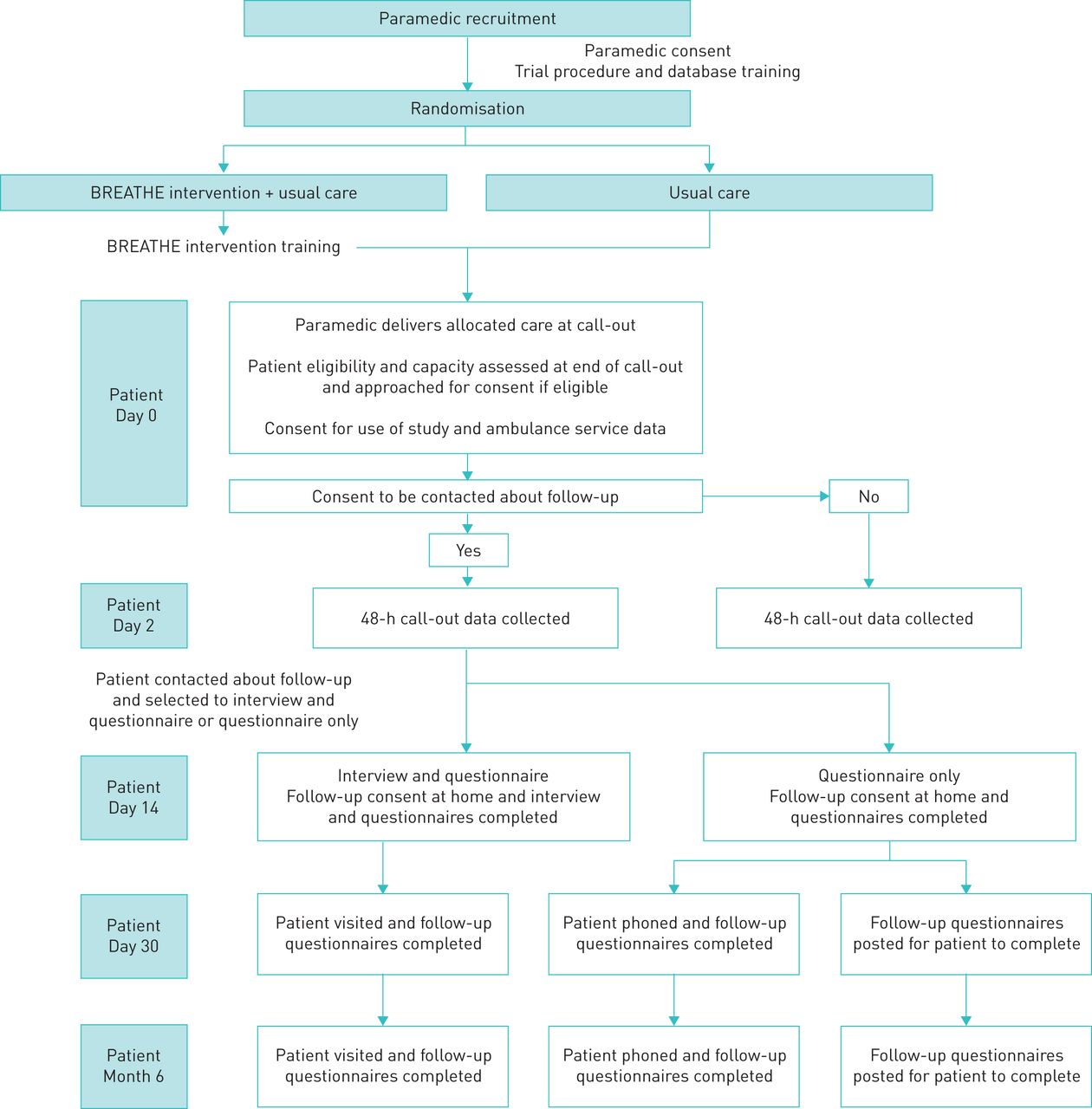
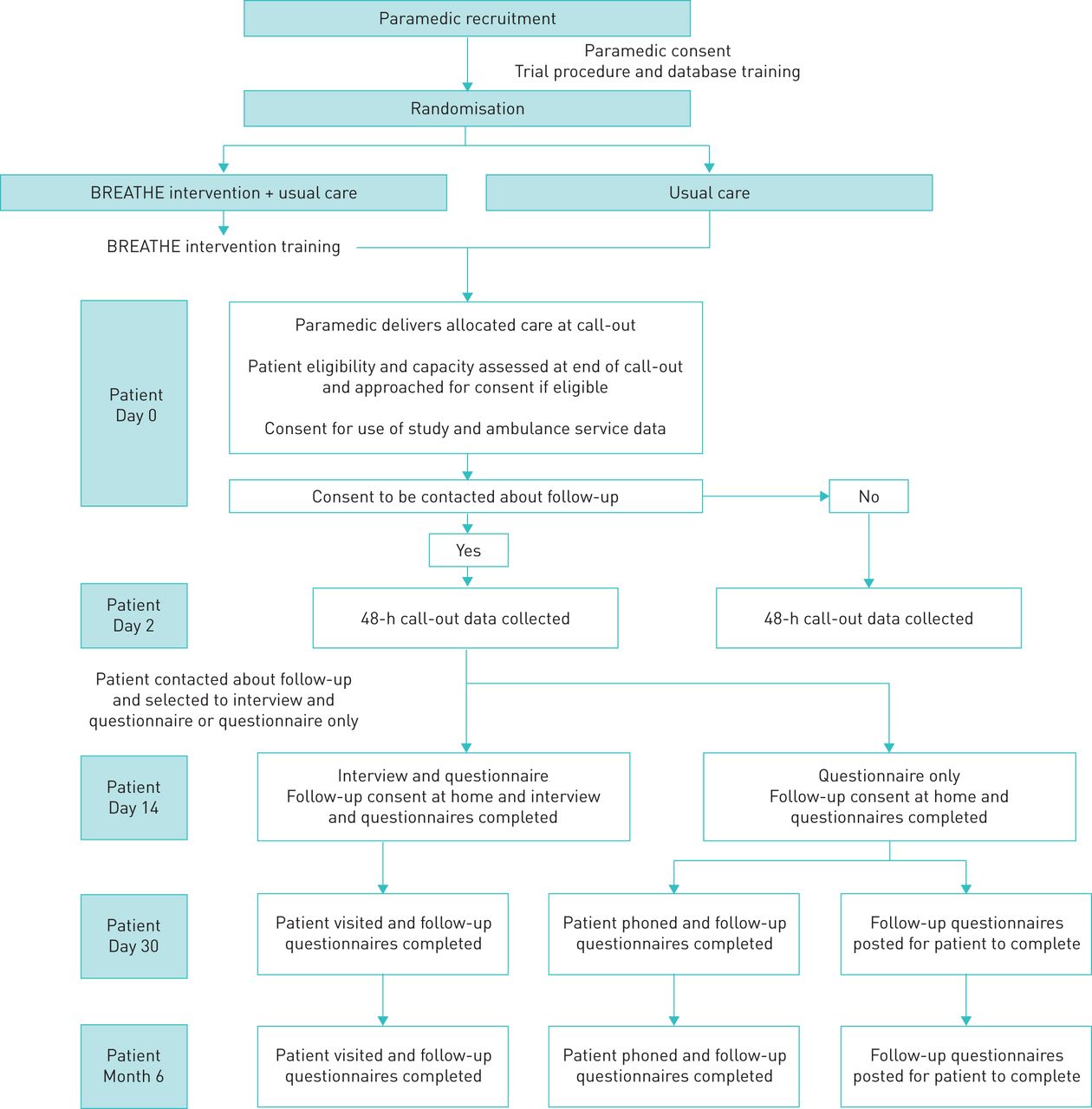{kind=link}
BREATHE Study flowchart.
Schedule of events for patient-participants
Study population
Paramedic-participants will be recruited from ambulance stations in one geographical region through advertisement and will be willing to undergo training in study measures, processes and the BREATHE intervention as required. All paramedics in the service are trained in the clinical assessment of the acutely unwell patient, with expertise in making hospital conveyance decisions either immediately or at the end of their visit in accordance with nationally agreed Joint Royal College Ambulance Liaison Committee (JRCALC) guidelines on initial assessment and oxygen use [21].
Eligible patients will be in their normal home environment receiving a 999 ambulance response from participating paramedics because of breathlessness. They will: have a self-reported diagnosis of a cardiorespiratory disease (including intra-thoracic malignancy); experience chronic breathlessness (defined as short of breath most days in the last 3 months or longer); and be able to give retrospective consent at the end of the call-out. Patients needing immediate life-saving intervention/transfer to the ED in the paramedic's clinical judgement are ineligible. They will also be excluded if they are currently enrolled on the trial or have previously participated. Carers present at call-out of any patient-participants consenting to be approached by the study team will be invited to take part in an interview.
Recruitment and consent
Paramedic-participants
The first eight paramedics to confirm a willingness to participate having read the paramedic-participant information sheet (PIS) will be invited to attend study initiation training. In the event of paramedic withdrawal prior to the training session, a paramedic from the waiting list will be invited. At the beginning of the training session an opportunity to ask any questions will be provided prior to being consented into the trial. Those randomised to BREATHE intervention plus usual care will also receive intervention training.
Patient-participants
Patient-participants will be identified and screened by participating paramedic-participants during the call-out. A two-step process will be adopted to patient-participant recruitment. From the beginning of the call-out, all paramedics will start recording the numerical rating scale (NRS) values of breathlessness severity. Paramedics randomised to deliver the BREATHE intervention will make an initial assessment of eligibility on arrival and deliver the intervention to those considered appropriate. At the end of the visit, whether staying at home, or being transferred, patients will be screened for eligibility, given the patient PIS and invited to participate. If willing, immediate written or witnessed verbal retrospective consent will be taken to use routinely collected clinical data from the index visit (Day 0), including the NRS measures, and details of any further call-outs in the subsequent 48 h.
Patient-participants can opt to be contacted about participation in further follow-up. Those agreeing to contact will be phoned by a member of the research team to discuss the purpose and processes of the follow-up and have the follow-up interview/questionnaires arranged on Day 14 (±7 days). In the event of admission to hospital at the initial visit, this would occur at the first opportunity after discharge home (maximum by Day 21). The patient PIS will be given to the patient-participant (and carer-participant if present) at the time of the visit and reviewed together to allow time to ask further questions before consent is taken.
Randomisation and blinding
Following consent, paramedics will be randomly allocated (1:1, random permuted blocks) using a commercial web-based randomisation system (REDCap Cloud) to BREATHE intervention plus usual care or usual care only.
Blinding is not possible for patient or paramedic-participants, or for members of the research team providing intervention training, as data will be collected at interview about intervention adherence and fidelity, and it is likely that patient-participants will indicate their allocation. The researcher conducting interviews will collect follow-up questionnaire data before conducting the interview in order to maximise the chances of remaining blinded to allocation until that point. After the initial follow-up visit, the researcher will note their guess of the allocation to check if blinding is possible to take forward into the full trial. The researcher collecting follow-up data but not interviewing participants will remain blinded to allocation. Researchers involved in the analysis of the quantitative data will be blinded to allocation.
Intervention and comparator
This study does not interfere with routine clinical decision-making, which will be conducted according to the JRCALC guidelines [21]. All clinical decisions regarding place of care will be taken according to the paramedic usual practice; highly experienced professionals who make decisions regarding conveyance daily.
Training
All paramedic-participants will receive a 1-h face-to-face study initiation training on consent and research procedures. This will be delivered by both clinical and non-clinical members of the research team and will include NRS training. The NRS breathlessness severity rating [22] (0=no breathlessness now; 10=worst possible breathlessness now) will form part of clinical assessment for all patients calling with acute-on-chronic breathlessness, irrespective of the trial. This is in line with recent calls to measure breathlessness severity as routine clinical practice. Paramedic-participants randomised to intervention only will receive face-to-face BREATHE intervention training delivered by clinicians on the research team.
Intervention
The BREATHE intervention is reported in accordance with the template for intervention description and replication (TIDieR) checklist [23]. The intervention (table 2) was developed based on components of evidence-based non-pharmacological breathlessness interventions [24] and the findings of in-depth interviews with respiratory and emergency clinicians. It includes: 1) face-to-face advice (positioning, breathing techniques, panic management, fan); 2) a laminated leaflet; and 3) a breathlessness management booklet to keep and refer to later. This booklet contains information to help the patient and carer self-manage breathlessness and information on local support services. Elements of the intervention used during the call-out will be recorded to capture fidelity of delivery.
BREATHE intervention and usual care
The BREATHE intervention has adapted everyday paramedic (first responders) practice aiming to ease acute-on-chronic breathlessness more quickly and, where the paramedic deems appropriate, prevent avoidable ED attendances in people living with chronic breathlessness. Following training, paramedic-participants allocated to intervention will use the intervention (or elements of it according to the clinical situation) during all call-outs for acute-on-chronic breathlessness irrespective of whether the patient consents to data collection.
Comparator: usual care
The paramedic-participants will deliver usual care according to national guidelines including initial history, baseline vital signs and tailored examination (chest auscultation, peak flow readings, 12-lead ECG).
Outcomes and assessments
For BREATHE, the primary end-point is the end of the index paramedic call-out. If the patient-participant stays at home, this will be at the point the paramedic leaves the house. If the patient is conveyed to the ED, this will be at any point up until the paramedic leaves the patient in the ED according to their clinical judgement.
Feasibility outcomes
The primary feasibility outcomes being assessed relate to recruitment. These are paramedic-participant and patient-participant recruitment and attrition overall, paramedics’ willingness to be randomised, patient recruitment per paramedic and consent for Day 0 data use, and for follow-up data provision.
The following secondary feasibility outcomes will be assessed:
Data quality: completeness of routinely collected data and of patient and proxy reported outcome measures.
Intervention: fidelity and adherence of the components delivered by the paramedics will be collected during the call-out. Acceptability will be assessed during patient and paramedic interviews. Whether the intervention continued to be used by the patient following the initial visit will be collected at 14 days, 1 month and 6 months.
Potential definitive trial primary outcomes
1) Improvement in breathlessness intensity at end of paramedic visit. NRS intensity every 2 min till decision to transfer to ED or decision to keep at home. The 0–10 NRS is a validated measure of breathlessness intensity [22], is highly correlated with Visual Analogue Scores, is more repeatable [33] and can be provided by a proxy reporter [34]. It can be used in routine clinical practice symptom assessment.
2) Conveyance to ED (from routinely collected data). The intra-cluster coefficient and sample size calculation for the candidate primary outcomes will be completed.
Clinical measures
Routinely collected clinical and service delivery data include pulse, respiratory rate, blood pressure, oxygen saturation measured by pulse oximetry (SpO2) with air, SpO2 with oxygen and working impression.
Health service utilisation
This includes healthcare resource use, both primary and secondary care, use of emergency services and self-reported use of community support services.
Health status
The SF-36 (Short Form 36) will be administered to patient-participants from which the SF-6D will be derived. The SF-6D is a validated health status measurement tool widely used in health economic evaluations [35].
Sample size
As a feasibility study, a formal sample size calculation has not been performed. Sixty patient-participants will be recruited over 6 months, 30 in each group, providing sufficient data to answer our feasibility and desirability questions [38]. Eight paramedic-participants allows for a 20% dropout rate, with the aim of including at least six paramedic-participants each treating 10 patient-participants, considered sufficient for calculating the intra-cluster coefficient.
Embedded NPT study
A mixed-methods approach will be used to conduct an embedded NPT [39] informed evaluation to explore barriers and enablers within implementing practice change domains: coherence, cognitive participation, collective action and reflexive monitoring. Semi-structured interviews will be held with a purposive sample of patient-participants (n=20) (including carer-participants where present). At the end of patient-participant recruitment, all paramedic-participants will be invited to participate in semi-structured interviews/focus groups (to explore views on study procedures/measures and issues regarding the implementation of the BREATHE intervention) and complete a NoMAD survey asking their opinion about whether and how the intervention could become part of routine clinical practice.
Data management
The main study database will be developed by Hull Health Trials Unit (HHTU), using the commercial electronic data capture system, REDCap Cloud. The system uses validation and verification features which will be used to monitor study data quality and completeness. A study monitoring plan will be developed by HHTU who will monitor the study.
Data analysis
The trial will be reported in accordance with the CONSORT 2010 statement extension to pilot and feasibility trials [40].
Descriptive statistics will be reported for the feasibility outcomes: paramedic recruitment rates by ambulance station, patient-participant recruitment rate by paramedic, intervention uptake, quality of data collection, intervention delivery and fidelity.
Baseline data and summary for candidate primary and secondary outcomes are summarised overall and by trial arm both by randomisation and separately for participants providing data to the primary end-point. Data will inform a potential definitive study in terms of patient/carer self-reported needs: NRS breathlessness intensity, clinical measures assessed by paramedics, health service utilisation questionnaire, SF-6D and mastery of breathlessness (CRQ). Variability in candidate primary measures will be calculated and a sample size (power calculation) for the definitive trial will be estimated for each. Adverse events will be summarised descriptively.
Missing data will be described but not imputed. No statistical comparisons between treatment groups will be undertaken on baseline or follow-up data as the trial is not designed to test effectiveness.
The quality of data collection for the SF-6D (derived from the SF-36) and health service utilisation information will be reported descriptively; however, quality-adjusted life-years (QALYs) will not be calculated.
Interview data will be analysed using framework analysis with reference to NPT [39] to determine acceptability and feasibility of a definitive trial and barriers and facilitators to implementation. Interview/focus group data will be analysed using NPT as a deductive framework whilst allowing for the inductive identification of themes.
NoMAD survey data will be presented using descriptive statistics.
Stop–go criteria
Stop–go criteria (table 3) will be used to assess the key feasibility objectives of recruitment and intervention adherence as to inform whether a main trial is possible and whether the design or other issues needs modification in order to conduct it successfully. Remediable barriers and their solutions will be identified from the NPT study. Process data will be used to describe interpreted timelines to identify “fixable”, “manageable” and “insurmountable” challenges to site opening, training, data collection and intervention fidelity with regard to both the future main trial and clinical implementation in the event of a positive trial.
Stop–go criteria
Ethics and dissemination
Regulatory approvals and trial oversight
The trial protocol has been reviewed and approved by the Yorkshire and The Humber–Sheffield Research Ethics Committee (REC reference: 19/YH/0314). The University of Hull is the study sponsor, and the HHTU is supporting trial delivery. A Trial Management Group has been convened to oversee trial delivery and operations. An independent Trial Steering Committee will provide overall supervision for the project on behalf of the Project Sponsor and Project Funder.
Safety considerations and adverse event reporting
Adverse event reporting is defined in the full study protocol. In the emergency clinical context, full safety reporting of adverse events (AEs) and serious adverse events (SAEs) will be limited to events that occur during the call-out visit. Because of the inherent limitation in collecting AE and SAE outcomes, proxy-safety data relating to the number and outcome of further ambulance call-outs in the 48-h period after the index visit will be collected using routinely collected data. In addition, the study will collect health resource utilisation information at the 14-day, 30-days and 6-month follow-up time points.
Dissemination
The study results will be disseminated to the appropriate stakeholders through presentations, conferences and peer-reviewed journals according to the BREATHE publication and dissemination policy.
Discussion
Providing it is found that delivering the BREATHE intervention is acceptable, feasible and desirable, the results will inform the number of paramedic clusters required, most appropriate primary outcome and the structure of a future definitive cluster randomised controlled trial in breathlessness patients.
Acknowledgements
We wish to acknowledge the contributions of Jane Shewan, Fiona Bell, Richard Pilbery, Elisha Miller and the participating paramedics from Yorkshire Ambulance Service. Susan Griffin (University of York, York, UK), Joanne Reeve (University of Hull, Hull, UK) and Pat Hatfield (PPI representative) for their input as co-applicants and members of the TMG. Finally, we thank Anne English (Dove House Hospice, Hull, UK) for her assistance in delivery of the paramedic training sessions.
Footnotes
This study is registered at www.isrctn.com with identifier number ISRCTN80330546. The main results paper will be submitted to a peer reviewed journal at study completion. It is intended that individual participant data that underlie the results reported in final results papers will be made available for sharing for research purposes after de-identification. Data will not be shared until after completion of the final study report to the funder but will then be available with no end date. Final anonymised clinical trial datasets and metadata will be produced in accordance with participant consent, and stored in an appropriate format to enable discoverability and sharing in the University of Hull's data repository HYDRA. Publications will include data availability statements.
Protocol version: Based on protocol version 2.0 (23 September 2019).
Authors contributions: A. Hutchinson, V. Allgar, S. Hart, A. Hodge, M. Johnson, S. Mason and F. Swan are coapplicants on the grant application. M. Northgraves, J. Cohen, V. Allgar, D. Currow, S. Hart, K. Hird, A. Hodge, M. Johnson, S. Mason, F. Swan and A. Hutchinson assisted in development of the protocol and implementation of the study. M. Northgraves, A. Hutchinson, M. Johnson and J. Cohen drafted the manuscript. All authors read and approved the final manuscript.
Conflict of interest: M. Northgraves has nothing to disclose.
Conflict of interest: J. Cohen reports grants from the NIHR during the conduct of the study.
Conflict of interest: V. Allgar has nothing to disclose.
Conflict of interest: D. Currow reports he is an unpaid advisory board member for Helsinn Pharmaceuticals. He is a paid consultant and receives payment for intellectual property with Mayne Pharma and is a consultant with Specialised Therapeutics Australia Pty. Ltd.
Conflict of interest: S. Hart reports personal fees and nonfinancial support from Chiesi UK; and grants, personal fees and nonfinancial support from Boehringer Ingelheim, all outside the submitted work.
Conflict of interest: K. Hird has nothing to disclose.
Conflict of interest: A. Hodge has nothing to disclose.
Conflict of interest: M. Johnson has nothing to disclose.
Conflict of interest: S. Mason has nothing to disclose.
Conflict of interest: F. Swan has nothing to disclose.
Conflict of interest: A. Hutchinson has nothing to disclose.
Support statement: This paper presents independent research funded by the National Institute for Health Research (NIHR) under its Research for Patient Benefit Programme (grant reference number PB-PG-0817-20009). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received December 18, 2020.
- Accepted January 7, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










