





Figures
- FIGURE 1
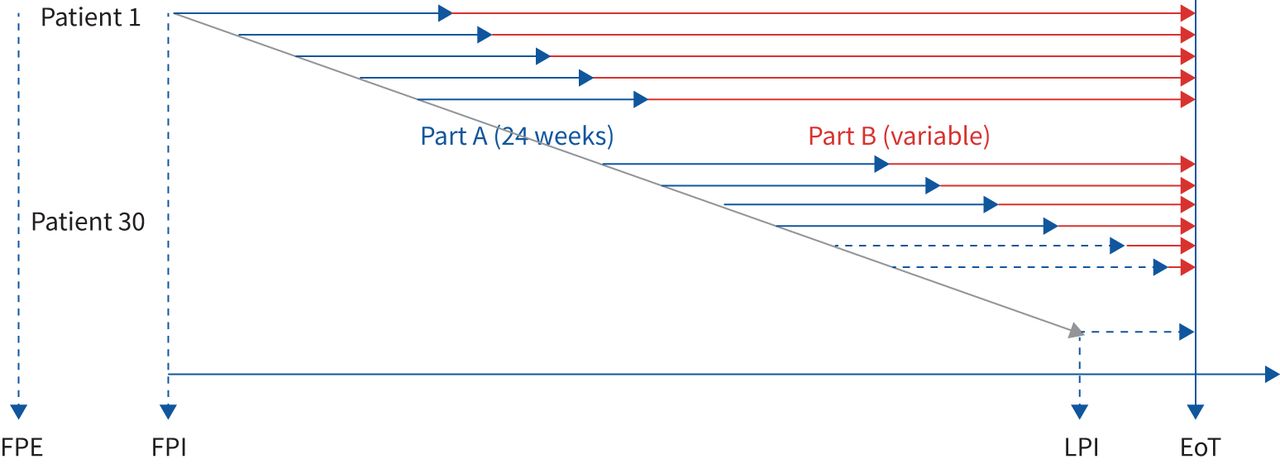
Study design. EoT: end of treatment; FPE: first patient enrolled; FPI: first patient in; LPI: last patient in.
- FIGURE 2
Inclusion criteria for fibrosing interstitial lung disease (ILD). Evidence of fibrosing ILD will be confirmed by central review (lung biopsy and high-resolution computed tomography (HRCT)). NSIP: non-specific interstitial pneumonia; UIP: usual interstitial pneumonia. #: Coexisting cystic abnormalities or ground-glass opacity are acceptable; however, coexisting multifocal non-fibrotic, non-dependent consolidations (e.g. organising pneumonia, infection) will not be permitted. ¶: With or without ground-glass opacification.
- FIGURE 3
Key assessments. #: Part A comprises visit 2 (week 0), visit 3 (week 2), visit 4 (week 6), visit 5 (week 12), and visit 6 (week 24). Laboratory tests can be performed between visit 5 and 6 (visit 5A) as needed. ¶: Part B starts at the end of visit 6 and comprises visit 7 (week 26), visit 8 (week 36), visit 9 (week 52), visit X (week 64, then every 12 weeks until end of treatment (EoT)), and EoT. Laboratory tests can be performed between each visit (visit 7A, 8A, 9A and XA as needed). +: Clinically significant disease is assessed by the investigator based on any of the following: Fan score ≥3 or documented evidence of clinical progression over time (either 5–10% relative decline in forced vital capacity (FVC) % predicted accompanied by worsening symptoms, or a ≥10% relative decline in FVC% predicted, or increased fibrosis on high-resolution computed tomography (HRCT), or other measures of clinical worsening attributed to progressive lung disease (e.g. increased oxygen requirement, decreased diffusion capacity)). §: The primary endpoints are pharmacokinetics (area under the plasma concentration–time curve at steady state (AUCτ,ss)) based on sampling at steady state (weeks 2 and 26) and number (%) of patients with treatment-emergent adverse events (week 24). 6MWT: 6-minute walk test; DLCO: diffusing capacity of the lung for carbon monoxide; ECG: electrocardiogram; ILD: interstitial lung disease; PedsQL: Pediatric Quality of Life Questionnaire; PK: pharmacokinetics; SpO2: oxygen saturation measured by pulse oximetry.



{kind=link}
{kind=link}
{kind=link}
Tables
- TABLE 1
Full inclusion and exclusion criteria
Inclusion criteria Male or female children and adolescents aged 6–17 years old at visit 2 Written informed consent and assent (where applicable) prior to admission to the trial Female subjects of childbearing potential must confirm that sexual abstinence is standard practice and will be continued until 3 months after last drug intake, or agree to use a highly effective method of birth control from 28 days prior to initiation of study treatment, during treatment and until 3 months after last drug intake Evidence of fibrosing ILD# on HRCT within 12 months of visit 1 as assessed by the investigator and confirmed by central review FVC% predicted ≥25% at visit 2¶ Clinically significant disease at visit 2, as assessed by the investigator based on any of the following: Fan score ≥3 [10], or
Documented evidence of clinical progression over time based on either:
o 5–10% relative decline in FVC% predicted accompanied by worsening symptoms, or
o a ≥10% relative decline in FVC% predicted, or
o increased fibrosis on HRCT, or
o other measures of clinical worsening attributed to progressive lung disease (e.g. increased oxygen requirement, decreased diffusion capacity)
Exclusion criteria AST and/or ALT >1.5×ULN at visit 1+ Bilirubin >1.5×ULN at visit 1+ Creatinine clearance <30 mL·min−1 calculated by Schwartz formula at visit 1+ Patients with underlying chronic liver disease (Child Pugh A, B or C hepatic impairment) at visit 1 Previous treatment with nintedanib Other investigational therapy received within 1 month or 5 half-lives (whichever is shorter but ≥1 week) prior to visit 2 Significant PAH defined by any of the following: Previous clinical or echocardiographic evidence of significant right heart failure
History of right heart catheterisation showing a cardiac index ≤2 L·min−1·m−2
PAH requiring parenteral therapy with epoprostenol/treprostinil
Other clinically significant pulmonary abnormalities (investigator-assessed) Cardiovascular diseases (any of the following): Severe hypertension (uncontrolled with treatment), within 6 months of visit 1; uncontrolled hypertension is defined as:
o Children aged 6–≤12 years: ≥95th percentile+12 mmHg or ≥140/90 mmHg (whichever is lower) (systolic or diastolic blood pressure equal to or greater than the calculated target value)
o In adolescents aged 13–17 years: systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg
Myocardial infarction within 6 months of visit 1
Unstable cardiac angina within 6 months of visit 1
Bleeding risk, defined as any of the following: Known genetic predisposition to bleeding
Patients who require:
o Fibrinolysis, full-dose therapeutic anticoagulation (e.g. vitamin K antagonists, direct thrombin inhibitors, heparin, hirudin)
o High-dose antiplatelet therapy§
History of haemorrhagic CNS event within 12 months of visit 1
Any of the following within 3 months of visit 1:
o Haemoptysis or haematuria
o Active gastrointestinal bleeding or gastrointestinal ulcers
o Major injury or surgery (investigator-assessed)
Any of the following coagulation parameters at visit 1:
o INR >2
o Prolongation of PT by >1.5×ULN
o Prolongation of aPTT by >1.5×ULN
History of thrombotic event (including stroke and transient ischaemic attack) within 12 months of visit 1 Known hypersensitivity to the trial medication or its components (i.e. soya lecithin) Documented allergy to peanut or soya Other disease that may interfere with testing procedures or in the judgement of the investigator may interfere with trial participation or may put the patient at risk when participating in this trial Life expectancy for any concomitant disease other than ILD <2.5 years (investigator-assessed) Female patients who are pregnant, nursing, or who plan to become pregnant while in the trial Patients not able or willing to adhere to trial procedures, including intake of study medication Patients with any diagnosed growth disorder such as growth hormone deficiency or any genetic disorder that is associated with short stature (e.g. Turner syndrome, Noonan syndrome, Russell–Silver syndrome) and/or treatment with growth hormone therapy within 6 months before visit 2ƒ Patients <13.5 kg of weight at visit 1 (same threshold for male and female patients) ALT: alanine aminotransferase; aPTT: activated partial thromboplastin time; AST: aspartate aminotransferase; CNS: central nervous system; FVC: forced vital capacity; HRCT: high-resolution computed tomography; ILD: interstitial lung disease; INR: international normalised ratio; PAH: pulmonary arterial hypertension; PT: prothrombin time; ULN: upper limit of normal. #: Clinically significant fibrosing ILD will be confirmed based on documented evidence of fibrosing features on HRCT or lung biopsy, as defined in the Participants section of the Methods. ¶: predicted normal values will be calculated according to the Global Lung Initiative. +: laboratory parameters from visit 2 will only be available after randomisation and, if the result no longer satisfies the entry criteria, the investigator will decide whether the patient should remain on study drug. Abnormal laboratory parameters at visit 1 are allowed to be re-tested (once) if it is thought that there was a measurement error or if they are a result of a temporary and reversible medical condition (once that condition has resolved). §: prophylactic low-dose heparin or heparin flush as needed for maintenance of an indwelling intravenous device, as well as prophylactic use of antiplatelet therapy, are not prohibited. ƒ: patients with short stature considered by the investigator to be due to glucocorticoid therapy may be included.
- TABLE 2
Study endpoints
Primary endpoints: Pharmacokinetics: AUCτ,ss based on sampling at steady state (at week 2 and week 26) Number (%) of patients with treatment-emergent adverse events at week 24 Secondary endpoints: Number (%) of patients with treatment-emergent pathological findings of epiphyseal growth plate on imaging at week 24 and week 52# Number (%) of patients with treatment-emergent pathological findings on dental examination or imaging at week 24 and week 52# Number (%) of patients with treatment-emergent adverse events over the whole trial Change in height, sitting height, leg length from baseline at week 24, week 52#, week 76# and week 100# Change in FVC% predicted from baseline at week 24 and week 52# Absolute change from baseline in PedsQL at week 24 and week 52# Change in SpO2 on room air at rest from baseline at week 24 and week 52# Change in 6-min walk distance from baseline at week 24 and week 52# Patient acceptability based on the size of capsules at week 24 Patient acceptability based on the number of capsules at week 24 Time to first respiratory-related hospitalisation over the whole trial Time to first acute ILD exacerbation or death over the whole trial Time to death over the whole trial Further endpoints Number (%) of patients with increase/decrease in FVC% predicted (5–10%; >10%) at week 24 and week 52# Number (%) of patients with ≥4.4-point increase in PedsQL from baseline at week 24 and week 52# Number (%) of patients with >4% increase in SpO2 on room air from baseline at week 24 and week 52# Change in calculated Fan severity score from baseline at week 24 and week 52# Change in HAZ score from baseline at week 24 and week 52# Change in WAZ score from baseline at week 24 and week 52# Slope of HAZ over whole trial Slope of WAZ over whole trial Number of missed school days due to the disease under study at week 24 Absolute change from baseline in log-transformed CA-125 at week 24 and week 52 Pharmacokinetic endpoints at Visit 3 (week 2) and Visit 7 (week 26) Cmax,ss tmax,ss t1/2,ss CL/Fss Vz/Fss Cpre,ss Other parameters may be calculated as deemed appropriate. AUCτ,ss: area under the plasma concentration–time curve at steady state; CL/Fss: apparent clearance of the analyte in the plasma at steady state following extravascular multiple dose administration; Cmax,ss: maximum measured concentration of the analyte in plasma at steady state; Cpre,ss: pre-dose concentration of the analyte in plasma at steady state immediately before administration of the next dose; FVC: forced vital capacity; HAZ: height-for-age z-score; ILD: interstitial lung disease; PedsQL: Pediatric Quality of Life Questionnaire; SpO2: oxygen saturation measured by pulse oximetry; t1/2,ss: terminal half-life of the analyte in plasma at steady state; tmax,ss: time from dosing to maximum measured concentration of the analyte in plasma at steady state; Vz/Fss: apparent volume of distribution during the terminal phase λz at steady state following extravascular administration; WAZ: weight-for-age z-score. #: 52 weeks, 76 weeks and 100 weeks time points will not be available for all patients.
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00805-2020.SUPPLEMENT










