





Abstract
Background Dysregulation of tumour necrosis factor-α (TNF-α) signalling is implicated in neutrophilic asthma. TNF-α signalling involves membrane-bound and soluble ligand (TNF-α) and receptors (TNFRs); however, little is known about how these proteins are altered in asthma. We hypothesised that intercompartment-, immune cell- and/or asthma inflammatory phenotype-dependent regulation could relate to TNF dysregulation in neutrophilic asthma.
Methods Measurements were made in 45 adults with asthma (36 non-neutrophilic, 9 neutrophilic) and 8 non-asthma controls. Soluble TNF-α, TNF receptor 1 (TNFR1) and TNFR2 were quantified in plasma and sputum supernatant by ELISA, and membrane-bound TNF-α/TNFR1/TNFR2 measured on eosinophils, neutrophils, monocytes, and macrophages in blood and sputum by flow cytometry. Marker expression was compared between inflammatory phenotypes and compartments, and relationship of membrane-bound and soluble TNF markers and immune cell numbers tested by correlation.
Results Soluble sputum TNFR1 and TNFR2 were increased in neutrophilic versus non-neutrophilic asthma (p=0.010 and p=0.029). Membrane-bound TNF-α expression was upregulated on sputum versus blood monocytes, while TNFR1 and TNFR2 levels were reduced on airway versus blood monocytes and neutrophils. Soluble TNFR1 and TNFR2 in sputum significantly correlated with the number of airway monocytes (p=0.016, r=0.358 and p=0.029, r=0.327).
Conclusion Our results imply that increased sputum soluble TNF receptor levels observed in neutrophilic asthma relate to the increased recruitment of monocytes and neutrophils into the airways and their subsequent receptor shedding. Monocytes also increase TNF-α ligand expression in the airways. These results suggest an important contribution of airway monocytes to the altered inflammatory milieu in neutrophilic asthma.
Abstract
Monocytes modify the inflammatory milieu in neutrophilic asthma. Blood and sputum analysis reveal TNF pathway dysregulation relating to altered TNF-α ligand and receptor expression by monocytes and their increased airway recruitment in neutrophilic asthma. https://bit.ly/3onJ1s7
Introduction
Asthma is a heterogeneous respiratory disease, involving recruitment of immune cells to the airways and the pathological release of inflammatory mediators [1]. Chronic inflammation of the airways promotes airway remodelling and variable airflow limitation, causing characteristic asthma symptoms such as wheezing, cough and breathlessness [2]. Different inflammatory phenotypes have been described based on the relative abundance of eosinophils and neutrophils in the airways [3, 4]. Heterogeneity in airway inflammation and the underlying molecular pathways is associated with varying responses to asthma medications. Eosinophilic asthma is promoted by release of Type 2 cytokines and is generally responsive to inhaled corticosteroids (ICS). Biologics targeting Type 2 cytokine signalling provide further clinical benefit in severe eosinophilic asthma [5]. Neutrophilic asthma is often refractory to ICS treatment [6], and targeted medication is lacking. Neutrophilic asthma is associated with more frequent exacerbations [7], airflow limitation [8] and increased bacterial burden [9], highlighting the need for more effective medication to treat neutrophilic asthma. A better understanding of the mechanisms underlying neutrophilic airway inflammation is crucial to identify novel therapeutic targets.
The tumour necrosis factor-α (TNF-α) signalling pathway plays an important role in immune responses, and its dysregulation is implicated in auto-inflammatory diseases including asthma. Recently, an upregulation of the TNF-α axis in neutrophilic asthma has been reported. Two transcriptomic studies observed increased expression of several genes related to TNF-α signalling in sputum of asthma participants with neutrophilic airway inflammation [10, 11]. TNF-α is a pro-inflammatory cytokine that interacts with the two receptors TNF receptor 1 (TNFR1) and TNFR2. TNFR1 either induces the expression of pro-inflammatory genes or initiates cell death, a process triggered by the intracellular death domain of TNFR1. TNFR2-induced signalling regulates the expression of genes related to cell proliferation and survival, homeostasis and anti-inflammatory processes [12–14]. TNF-α and TNFRs are expressed as transmembrane proteins that can be cleaved and become soluble, which has important regulatory functions [15, 16]. Both membrane-bound and soluble TNF-α activate TNFR1, whereas TNFR2 requires the membrane-bound variant to become fully activated. Receptor shedding can occur following ligation and is thought to terminate TNF-α-induced signalling by desensitising the cell to the ligand, and by sequestering soluble (s)TNF-α [17, 18]. In asthma, soluble sputum TNF-α, TNFR1 and TNFR2 are increased during exacerbations [19]. We recently demonstrated that increased sTNFR levels in sputum were associated with neutrophilic and severe asthma, poorer lung function and worse asthma control [20]. Therapeutic blockade of TNF-α has been shown to reduce inflammation in other chronic diseases such as rheumatoid arthritis [21]. In asthma, clinical trials of TNF-α-blocking biological therapies have produced varying outcomes [22]. Long-term azithromycin treatment reduces airway and circulating TNF-α and/or TNFRs in moderate–severe asthma [20]. Better characterisation of the heterogeneous dysregulation of TNF-α signalling is required to understand the potential of therapeutically targeting the TNF-α pathway in asthma.
Here we aimed to understand how TNF-α ligand and receptor expression and/or shedding are dysregulated in asthma. We hypothesised that TNF marker levels would be altered in neutrophilic asthma. Based on previous reports [20], we expected increased sTNFR levels in sputum of participants with neutrophilic asthma. We further hypothesised that membrane-bound TNF marker expression would vary across immune cell types, compartments and inflammatory phenotypes. Lastly, we hypothesised that soluble sputum TNF marker levels would correlate with their membrane-bound variants and/or the number of immune cells in the airways. To test our hypotheses, we quantified soluble TNF-α, TNFR1 and TNFR2 in plasma and sputum supernatant by ELISA and performed comparisons between neutrophilic asthma versus non-neutrophilic asthma versus non-asthma controls. Within the same cohort we measured membrane-bound TNF markers on blood and sputum immune cells by flow cytometry. We identified those cell types that express detectable amounts of TNF-α/TNFRs and investigated whether their expression levels differed between groups or between compartments. Lastly, we explored the relationship between soluble marker levels and their membrane-bound counterparts and between soluble marker levels and sputum immune cell numbers.
Methods
Participants
Study measures were made in a published cohort of participants with asthma (n=45) and non-asthma controls (n=8) [23]. Participants were aged ≥18 years and provided written informed consent. Clinical studies were approved by the Hunter New England Human Research Ethics Committee and the University of Newcastle Human Research Ethics Committee (approval numbers: 16/04/20/3.0; 17/02/15/3.04; 15/03/18/3.04; 17/04/12/4.03). Participants with asthma had a doctor diagnosis of asthma with variable airflow obstruction (see online supplementary material). Information on demographics, asthma history, medication use and adherence, asthma symptoms based on Juniper Asthma Control Questionnaire 6 (Acq6 [24]), exacerbation history and smoking history was collected. Non-asthma control participants were eligible if they were not pregnant or breastfeeding, had not smoked within the last 6 months and did not have a current respiratory disease.
Spirometry, sample collection and processing
Spirometry, sputum induction and sample processing were performed as previously described [25] (see online supplementary material). Pre-bronchodilator forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC) and FEV1/FEV were measured (Medgraphics CPFS/D USB, Ascencia). Participants with asthma were classified as neutrophilic (≥61% neutrophils) or non-neutrophilic (<61% neutrophils) based on sputum differential cell counts and cut-offs [3, 4]. Venous blood samples were collected, and an automated full blood count was performed (CELL-DYN Ruby, Abbott, Chicago, IL, USA).
Flow cytometry of whole blood and sputum
Staining was performed as outlined in the online supplementary material. 100 µL whole blood and 1×105–1.5×106 sputum cells were labelled with antibodies directed against CD45, CD14, CD16, HLA-DR, TNF-α, TNFR1 and TNFR2 (see online supplementary material for details). Sputum cells were further stained with anti-CD206 and a viability dye (7-AAD). Isotype controls for TNF-α, TNFR1 and TNFR2 (BD Biosciences) were tested in a subset of sputum and blood samples. Samples were analysed on a flow cytometer (LSRFortessa X-20, BD Biosciences) and subsequent analyses performed with the software FlowJo (Version 10).
Gating of immune cells and quantification of membrane-bound TNF proteins
Monocytes, neutrophils and eosinophils in sputum and blood, and macrophages in sputum were identified based on the gating strategy described previously [23] and as illustrated in supplementary figures S1 and S2. We further distinguished monocyte subsets based on CD14/CD16 expression: classical (CM: CD14++CD16−), intermediate (IM: CD14+CD16+) and non-classical monocytes (NCM: CD14+CD16++; supplementary figure S3 [23]). TNF-α, TNFR1 and TNFR2 expression on immune cells in blood and sputum were quantified by determining the median fluorescence intensity (MFI) of their conjugated fluorochromes and subtracting the mean MFI of the corresponding isotype controls measured on the same cell type in the same compartment.
Enzyme-linked immunosorbent assays
ELISA kits (Human TNFα Quantikine High Sensitivity Kit; Human TNFR1/TNFRSF1A DuoSet; Human TNFRII/TNFRSF1B DuoSet) were purchased from R&D Research and TNF markers measured following manufacturer's instructions as previously published [20]. 1:20 dilutions of plasma were used for the detection of TNFR1 and TNFR2. TNF-α was measured in neat plasma samples. 1:5 dilutions of sputum supernatant were used for all three TNF measures.
Statistical analysis
Statistical analyses were performed using Stata 15 and GraphPad Prism 8. Normality and lognormal distribution were assessed using the Shapiro–Wilk test. Kruskal–Wallis with Dunn's correction was used for multiple comparison of non-parametric data and the one-way ANOVA test for normally distributed data. Wilcoxon matched-pairs signed rank test was used for the comparison of matched samples between blood and sputum. Correlation analyses were performed using Spearman's rank for non-parametric data.
Results
Participant characteristics
Characteristics of the participants are summarised in table 1. The non-neutrophilic asthma group had a significantly higher body mass index and a higher proportion of sputum eosinophils than the non-asthma control group. Neutrophilic asthma was associated with significantly higher total sputum cell count, neutrophil proportion and number, and a reduced proportion of macrophages compared to the other groups.
Demographics and clinical characteristics of the study population
Neutrophilic asthma features increased soluble sputum receptors
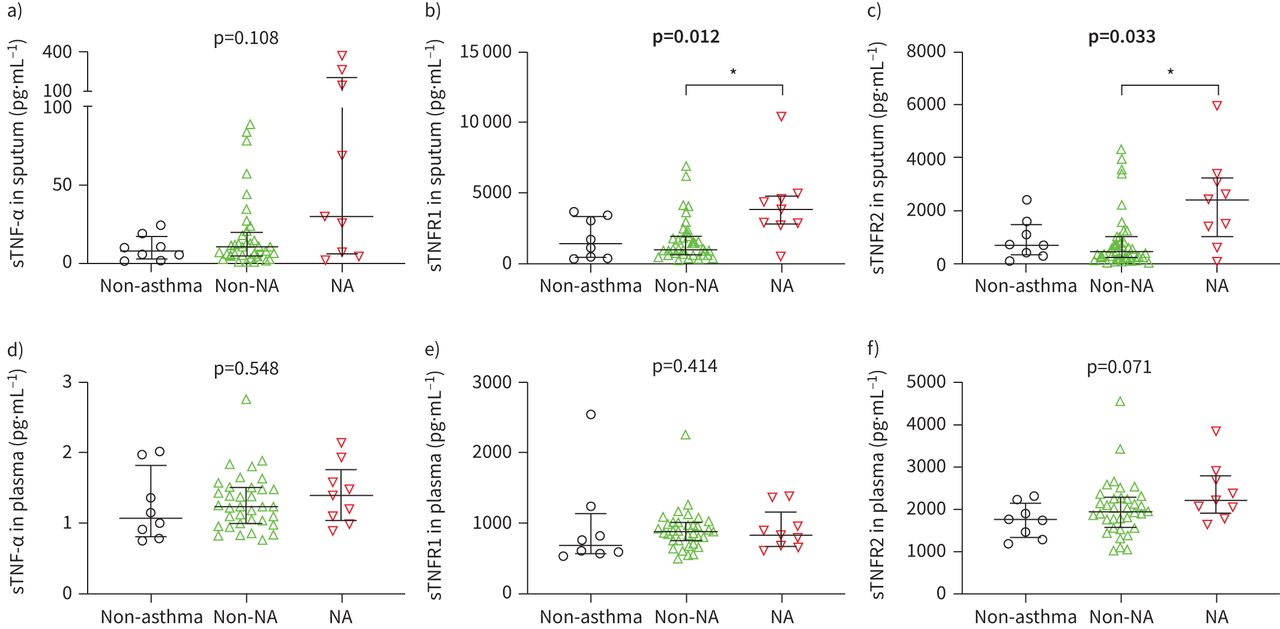
We quantified soluble variants of TNF markers in sputum supernatant and plasma by ELISA and performed comparisons between neutrophilic asthma, non-neutrophilic asthma and non-asthma controls. Sputum sTNF-α levels did not differ significantly between groups (figure 1a). Neutrophilic asthma featured significantly higher levels of soluble sputum TNFR1 (p=0.010; figure 1b) and TNFR2 (p=0.029; figure 1c). We observed no differences in sTNF marker levels in plasma (figure 1d–f).

Comparison of soluble (s) tumour necrosis factor (TNF)-α, TNF receptor (TNFR)1 and TNFR2 in sputum supernatant (a–c) and plasma (d–f) between non-asthma controls (black, n=8), non-neutrophilic asthma (non-NA) (green, nplasma=35, nsputum=36) and neutrophilic asthma (NA) (red, n=9). Bars and whiskers represent the median and the interquartile range. p-values were derived from the Kruskal–Wallis test. Significant p-values are highlighted in bold. *: p<0.05 level for pairwise comparisons (Dunn's).
Surface expression of TNF markers varies across immune cells
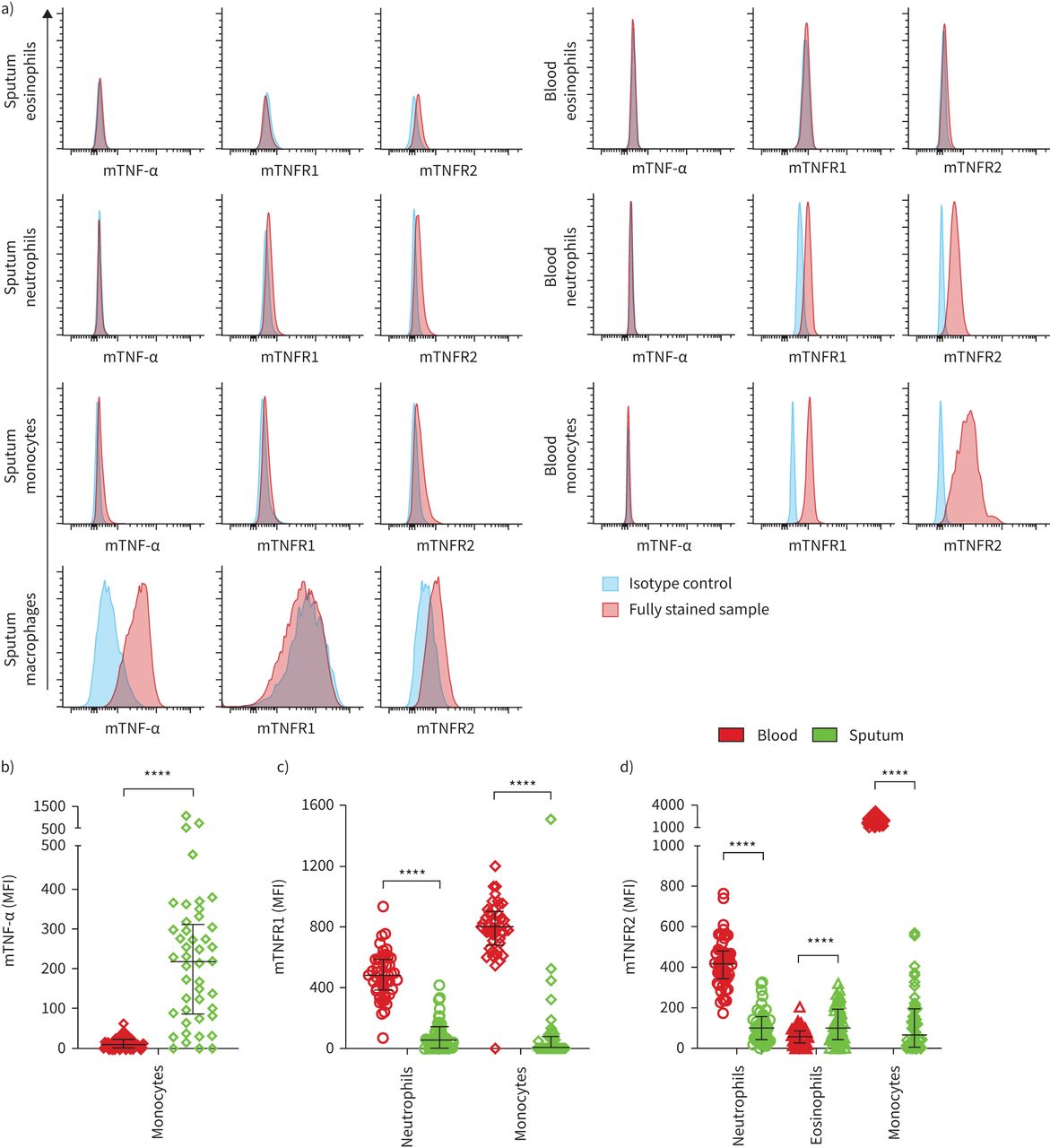
We quantified the relative expression levels of membrane-bound TNF-α, TNFR1 and TNFR2 on eosinophils, neutrophils, monocytes in blood and sputum and on macrophages in sputum using flow cytometry. Isotype controls were used to detect unspecific staining and to assess which cell types had detectable levels of the proteins of interest. Representative isotype controls and fully stained sputum and blood samples are illustrated in figure 2a.

Surface expression of membrane-bound (m) tumour necrosis factor (TNF)-α, TNF receptor (TNFR)1 and TNFR2 on immune cells in the circulation and the airways. a) Fluorescence intensities of fully stained samples and the corresponding isotype control in sputum (left) and blood (right). Comparison of b) mTNF-α, c) mTNFR1 and d) mTNFR2 expression levels on blood and sputum immune cells in asthma (n=45). Bars and whiskers indicate the median with interquartile range. The Wilcoxon matched-pairs signed rank test was used for statistical comparison. MFI: median fluorescence intensity. ****: p<0.0001.
On eosinophils we detected low but measurable levels of TNFR2 in blood and sputum. Neutrophils showed a distinct positive signal for TNFR1 and TNFR2 in both compartments. All three proteins were detected on monocytes in blood and sputum, although TNFR1 was undetectable on sputum monocytes in most samples and excluded from further analyses. Sputum macrophages showed a distinct positive signal for TNF-α and TNFR2. The fluorescence intensities of TNF-α and TNFR1 on eosinophils, TNF-α on neutrophils and TNFR1 on macrophages were similar to the fluorescence intensity of the corresponding isotype control, suggesting no or low expression of these markers on these cell types, and these marker/cell combinations were not included in further analyses.
TNF marker surface expression differs between compartments
We compared the expression levels of membrane-bound TNF markers on blood versus sputum immune cells in participants with asthma, excluding those cell type/marker combinations in which expression was undetectable in both compartments (figure 2b–d). TNF-α showed low detection levels on blood monocytes, whereas sputum monocytes featured higher levels of TNF-α. TNFR1 and TNFR2 on neutrophils and monocytes were more abundant in the circulation compared to the airways. Eosinophils featured lower TNFR2 expression in blood than in sputum. We observed similar differences in the expression patterns between compartments on monocytes and neutrophils in the non-asthma control group, while TNFR2 expression levels on eosinophils did not differ significantly between blood and sputum (supplementary figure S4).
Blood monocyte subsets feature distinct TNF marker expression patterns
We compared TNF marker expression levels on monocyte subsets within our asthma cohort and found no differences between subsets in sputum (supplementary figure S5a–c). In blood, we observed significantly higher TNF-α levels on intermediate (IM) versus non-classical monocytes (NCM) (p=0.009; supplementary figure S5d). mTNFR1 was significantly higher on classical monocytes (CM) and IM compared to NCM (p<0.001; supplementary figure S5e), whereas mTNFR2 inversely increased significantly from CM to IM to NCM (p<0.001; supplementary figure S5f). These findings were similar to those in the non-asthma cohort (data not shown).
Surface marker expression on airway immune cells is not altered in NA
We hypothesised that increased shedding of membrane-bound TNF markers in neutrophilic asthma may promote the increases in soluble TNF markers and would be reflected by lower detectable surface expression. However, there were no differences in surface expression levels of TNF markers on sputum immune cells between neutrophilic asthma versus non-neutrophilic asthma versus non-asthma controls (figure 3).

{kind=link}
{kind=link}
{kind=link}
Surface expression of membrane-bound (m) a) and b) tumour necrosis factor (TNF)-α, c) TNF receptor (TNFR)1 and d–g) mTNFR2 on sputum immune cells in neutrophilic asthma (NA), non-neutrophilic asthma (non-NA) and non-asthma controls. Non-asthma, n=8; non-NA, n=36; NA, n=9. p-values were derived from the Kruskal–Wallis test unless specified otherwise. Bars and whiskers indicate the median with the interquartile range for non-parametric data, and mean±sd for parametric data (b). MFI: median fluorescence intensity.
We further investigated the association between membrane-bound and soluble TNF marker levels in sputum (table 2). There was no significant correlation between sTNF-α and mTNF-α and no association between sTNFR1 and mTNFR1. Soluble sputum TNFR2 significantly positively correlated with mTNFR2 on sputum eosinophils (p=0.004, r=0.426) and sputum macrophages (p=0.017, r=0.353).
Spearman correlation matrix between soluble (s) tumour necrosis factor (TNF) marker levels in sputum supernatant and membrane-bound counterparts on sputum immune cells
We also tested if the difference between surface marker levels in blood and sputum correlated with sTNF sputum marker levels (table 3). We observed a significant negative correlation between the difference in mTNFR2 levels on eosinophils and sputum sTNFR2, signifying that an increased expression of mTNFR2 in sputum relative to blood related to increased levels of sTNFR2 in sputum.
Spearman correlation matrix of the difference (Δ) between membrane-bound (m) tumour necrosis factor (TNF) marker levels between blood and sputum with soluble (s) variants in sputum supernatant
Soluble sputum TNFR levels correlate with sputum monocyte numbers
We hypothesised that elevated soluble TNF marker levels in neutrophilic asthma sputum may be related to increased recruitment of immune cells to the airways; thus we tested the association between sputum immune cell numbers and soluble TNF marker levels (table 4). Absolute numbers of sputum monocytes correlated positively with sputum sTNFR1 (p=0.016, r=0.358) and sTNFR2 (p=0.029, r=0.327). No other significant correlations were detected.
Correlation between absolute numbers of sputum immune cells and soluble (s) tumour necrosis factor (TNF) variants in sputum supernatant
Discussion
TNF-α signalling regulates important pro- and anti-inflammatory functions, cellular communication, cell survival and death. Thus, aberrant TNF-α signalling is implicated in the onset of various inflammatory diseases. In our study we aimed to understand how the expression and cleavage of TNF-α, TNFR1 and TNFR2 are altered in asthma, particularly in relation to neutrophilic airway inflammation. Neutrophilic asthma was associated with significantly increased levels of sTNFR1 and sTNFR2 in sputum, validating our previous observation in the AMAZES cohort [20]. We demonstrate for the first time that the surface expression of TNF markers is both immune cell and compartment dependent. Monocyte expression of mTNF-α is increased in sputum versus blood, while reduced expression of both receptors on sputum versus blood monocytes and neutrophils implies these cells shed TNFRs in the airways. The three different monocyte subsets featured distinct TNF marker expression patterns in blood, suggesting different functions. In sputum, however, we did not observe any differences. Of the immune cells tested, only sputum monocyte number correlated with sTNFR levels. Macrophages expressed high levels of membrane-bound TNF-α that was not altered in neutrophilic asthma but were devoid of TNFR1 expression. Overall, our data suggest an important contribution of increased recruited monocytes to the altered airway inflammatory environment and TNF-α pathway dysregulation in neutrophilic asthma.
Neutrophilic asthma features dysregulation of the TNF-α signalling pathway at a transcriptional level. TNFR2 mRNA is increased in sputum of participants with neutrophilic airway inflammation [10, 11]. Based on these results, we anticipated increased expression levels of mTNFR2 on sputum immune cells in neutrophilic asthma. Unexpectedly, we did not observe any differences in membrane-bound TNF marker levels between neutrophilic asthma and non-neutrophilic asthma, arguing against aberrant expression or shedding of these proteins in any of the studied cell types in neutrophilic asthma. Yet, sputum sTNFR1 and sTNFR2 were significantly increased in neutrophilic asthma versus non-neutrophilic asthma. We performed correlation analyses between membrane-bound and soluble TNF marker levels to investigate where soluble receptors stem from, i.e., whether they are released from a specific cell type. Although sputum sTNFR2 correlated with mTNFR2 on sputum eosinophils and macrophages, these findings probably do not explain elevated sputum sTNFR2 in neutrophilic asthma, as eosinophil and macrophage numbers are decreased in neutrophilic asthma [23]. We observed a significant positive correlation between sputum sTNFRs and absolute numbers of sputum monocytes. This finding is of particular interest, as we previously observed increased recruitment of monocytes to the airways in neutrophilic asthma [23]. The significantly lower mTNFR levels found on sputum versus blood monocytes are suggestive of receptor shedding during or post transit into the airways. This reduction in mTNFR levels on monocytes (and neutrophils) in sputum was also observed in non-asthma controls, suggesting this is not a disease-specific phenomenon. Decreased mTNFR levels observed on sputum neutrophils and monocytes could also relate to increased internalisation of surface markers or increased exosome release from these cells in the airway lumen. Together, our findings support the hypothesis that the increased soluble sputum TNFR levels in neutrophilic asthma result from increased monocyte recruitment to the neutrophil-enriched airways, and subsequent shedding of monocyte TNFRs upon transit to the airways. The correlation reported is modest and suggests other cell types could play a role. Neutrophils showed a similar pattern of lower mTNFR in sputum versus blood and could also contribute, although their numbers in sputum were not significantly correlated with sTNFR levels. The potential contribution of the airway epithelium remains untested. Increased sputum sTNFR levels are related to more severe asthma, more frequent exacerbations, reduced lung function and asthma control [20]. These findings highlight the importance of following up transcriptomic studies with protein measures and suggest an important contribution of monocytes to the altered airway inflammatory milieu in neutrophilic asthma.
Interestingly, we observed significantly different TNF receptor levels on blood monocyte subsets. mTNFR1 levels decreased from CM to IM to NCM, whereas mTNFR2 inversely increased, suggesting that the different monocyte subsets favour distinct TNF-α signalling pathways, possibly associated with their functions. The observation that mTNF marker levels expressed on sputum monocytes did not differ across subsets suggests that increased TNF-α expression and TNF receptor shedding or downregulation through internalisation is similarly induced in the airways irrespective of the monocyte subset.
Soluble TNF-α ligand levels did not differ significantly between neutrophilic asthma, non-neutrophilic asthma and non-asthma controls in either compartment, although there was a trend towards increased sputum sTNF-α in neutrophilic asthma similar to that observed in the AMAZES cohort [20]. Other studies observed a significant increase in sTNF-α in serum and bronchoalveolar lavage fluid in participants with uncontrolled, severe asthma compared to mild asthma and healthy controls [26, 27], suggesting that increased TNF-α expression and shedding is more prevalent in severe asthma. Interestingly, neutrophils did not appear to express detectable amounts of mTNF-α in either compartment, implying that they are not a significant source of sTNF-α in the airways. We measured detectable and relatively high levels of mTNF-α on sputum monocytes and macrophages, respectively, which have been implicated as important producers of TNF-α in prior studies [28]. This finding is also consistent with their proposed immunoregulatory and homeostatic functions in the airways. Soluble TNF-α is rapidly released during infections, contributing to inflammation and immune cell recruitment, whereas mTNF-α expressed on macrophages induces neutrophil apoptosis via cell-to-cell interaction [29, 30]. In contrast to mTNF-α, we did not observe any detectable amounts of mTNFR1 on sputum macrophages. This observation was unexpected as macrophages in other compartments and in vitro have been reported to express both receptors [12], and crucial functions such as macrophage activation and cell death are linked to TNFR1-induced signalling [29]. Macrophages adopt unique tissue-specific phenotypes informed by local environmental cues, and thus it is possible that sputum macrophages adopt a phenotype lacking TNFR1. This is, to our knowledge, the first characterisation of macrophage mTNFR expression in sputum, and confirmation in independent studies will be required. The observed expression profile on macrophages with high levels of mTNF-α and mTNFR2 and no mTNFR1 might reflect a preferential signalling of airway macrophages through TNFR2 consistent with their proposed homeostatic roles in the airways.
Our study has some limitations. Only samples with sufficient sputum cell numbers have been used for analyses, which might have introduced bias. Owing to limited material for staining, we were able to include isotype controls only in a small subset of samples. Therefore, we subtracted the MFI of those isotype controls from the fluorescence intensity of the corresponding protein of interest and cell type across all samples. Although we ensured our flow cytometer passed quality control tests prior to each sample and our measures of surface fluorescence appeared consistent, we did not employ a standardised biological sample control across all samples so cannot exclude the possibility that unaccounted fluctuations in signal of measured proteins may have occurred over the course of the study. We cannot exclude that the presence of dithiothreitol during preparation of sputum cell suspensions may have had a differential effect on surface TNF marker expression compared to blood samples [31]. Our sputum flow cytometry gating strategy was conservative, and it is possible we may have omitted some cells from analysis, although prior cell sorting confirmed the accuracy of gating for macrophages, neutrophils, monocytes and eosinophils [23]. The sample sizes of the neutrophilic asthma and the non-asthma control groups were relatively small and might have limited the statistical power in detecting differences between groups. A potential contribution of sTNF markers derived from immune cells other than the investigated cell types should be investigated in further studies. Regardless of these limitations, this study provides detailed characterisation of the balance between blood and airway, membrane-bound and soluble TNF-α ligand and receptors in asthma for the first time.
In summary, our study demonstrates significant alterations of the inflammatory milieu in the airways in neutrophilic asthma. We confirmed prior reports that sTNFRs are increased in the sputum of participants with neutrophilic asthma. This coincided with significantly lower expression levels of mTNFRs on sputum monocytes and neutrophils compared to those cells in the circulation, suggesting that these receptors are cleaved and shed from these cells upon transit to the airways. The amount of membrane-bound TNF-α and TNFRs did not differ between neutrophilic asthma and non-neutrophilic asthma, and increased sTNFRs in sputum were related to the increased numbers of monocytes present in neutrophilic asthma sputum. The data suggest an important contribution of newly recruited monocytes to TNF-α dysregulation and the altered inflammatory environment in neutrophilic asthma.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00131-2021.SUPPLEMENT
Acknowledgements
The authors would like to acknowledge the support of the clinical and laboratory team members at HMRI who conducted participant recruitment, spirometry, and specimen collection, processing and analysis: Catherine Delahunty, Netsanet Negewo, Penelope Chan, Naomi Fibbens, Lakshitha Gunawardhana, Bridgette Donati, Heather Macdonald, Amber Smith and Allison Kautto. We would like to thank Nicole Cole (University of Newcastle) for assistance with flow cytometry and Collin Brooks (Massey University, New Zealand) for technical advice.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Author contributions: N.M. Niessen designed and conducted laboratory studies, designed and performed data analysis, interpreted the data, and drafted the manuscript. P.G. Gibson, J.L. Simpson and K.J. Baines designed and supervised clinical studies, supervised laboratory studies, designed data analysis, interpreted data and edited the manuscript. H.A. Scott supervised clinical studies, interpreted data and edited the manuscript. M. Fricker designed, conducted and supervised laboratory studies, designed data analysis, interpreted the data, and edited the manuscript.
Conflict of interests: N.M. Niessen has nothing to disclose.
Conflict of interest: P.G. Gibson reports, during the conduct of this study, that he holds a Practitioner Fellowship to the National Health and Medical Research Council. Outside the submitted works, grant funding has been received from AstraZeneca and GlaxoSmithKline, and he has participated in educational symposia funded by AstraZeneca, GlaxoSmithKline and Novartis.
Conflict of interest: J.L. Simpson has nothing to disclose.
Conflict of interest: H.A. Scott has nothing to disclose.
Conflict of interest: K.J. Baines reports project grant funds paid by NHMRC to their institution outside the submitted works.
Conflict of interest: M. Fricker reports receiving grant funding from GlaxoSmithKline outside the submitted works.
Support statement: This study was supported by the National Health and Medical Research Council (NHMRC) Centre of Research Excellence in Severe Asthma (project identifier 1078579), NHMRC project grants, Asthma Australia and the John Hunter Hospital Charitable Trust. M. Fricker was supported by a Thoracic Society of Australia and New Zealand-AstraZeneca Respiratory Research Fellowship. P.G. Gibson was supported by an NHMRC Practitioner Fellowship. K.J. Baines was supported by a Lung Foundation Australia COPD Research Fellowship. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received February 25, 2021.
- Accepted May 3, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










