





Abstract
Oxidative stress from innate immune cells is a driving mechanism that underlies COPD pathogenesis. Individuals with α-1 antitrypsin (AAT) deficiency (AATD) have a dramatically increased risk of developing COPD. To understand this further, the aim of this study was to investigate whether AATD presents with altered neutrophil NADPH oxidase activation, due to the specific lack of plasma AAT.
Experiments were performed using circulating neutrophils isolated from healthy controls and individuals with AATD. Superoxide anion (O2−) production was determined from the rate of reduction of cytochrome c. Quantification of membrane NADPH oxidase subunits was performed by mass spectrometry and Western blot analysis. The clinical significance of our in vitro findings was assessed in patients with AATD and severe COPD receiving intravenous AAT replacement therapy.
In vitro, AAT significantly inhibited O2− production by stimulated neutrophils and suppressed receptor stimulation of cyclic adenosine monophosphate and extracellular signal-regulated kinase (ERK)1/2 phosphorylation. In addition, AAT reduced plasma membrane translocation of cytosolic phox components of the NADPH oxidase. Ex vivo, AATD neutrophils demonstrated increased plasma membrane-associated p67phox and p47phox and significantly increased O2− production. The described variance in phox protein membrane assembly was resolved post-AAT augmentation therapy in vivo, the effects of which significantly reduced AATD neutrophil O2− production to that of healthy control cells.
These results expand our knowledge on the mechanism of neutrophil-driven airways disease associated with AATD. Therapeutic AAT augmentation modified neutrophil NADPH oxidase assembly and reactive oxygen species production, with implications for clinical use in conditions in which oxidative stress plays a pathogenic role.
Abstract
Circulating neutrophils in COPD due to α1-antitrypsin deficiency illustrate increased NADPH oxidase assembly and reactive oxygen species production, a defect corrected by α1-antitrypsin augmentation therapy https://bit.ly/38NNTzM
Introduction
α-1 antitrypsin (AAT) is a highly expressed serine protease inhibitor [1]. Hepatocytes are the main source of AAT [2], but it can also be synthesised by immune [3–5] and bronchial epithelial cells [6]. It is most abundant in plasma at a concentration of ∼1.5 g·L−1, and can diffuse into body fluids including tears, saliva and lung epithelial lining fluid (ELF) [7]. AAT's key function is to negate the proteolytic effects of neutrophil elastase and other serine proteases in the lung thus maintaining a protease–antiprotease balance [8]. Much of our understanding of the function of AAT comes from the study of individuals deficient in the protein. AAT deficiency (AATD) is an autosomal co-dominant condition caused by mutations in SERPINA1. The most common manifestations of AATD are early onset emphysema, yet progression of disease is understood to be a more complex scenario than the simple paradigm of a protease–antiprotease imbalance. Abnormal neutrophil and monocyte responses play a pivotal role in the progression of inflammation and lung disease in AATD [5, 9]. AAT has been shown to bind membrane lipid rafts [4], and localisation to such signalling platforms may in part explain its ability to modify essential neutrophil and monocyte functions including leukotriene B4-stimulated adhesion [10], cell migration to interleukin-8 (IL-8) [4], degranulation and proapoptotic signals [11, 12], and to regulate CD14 expression [13].
The only approved targeted therapy for individuals with AATD is weekly intravenous infusions of plasma purified AAT, often referred to as replacement therapy, administered at a standard dose of 60 mg·kg−1. This treatment increases plasma and ELF levels of AAT [7] and has been shown to reduce loss of lung tissue density [14, 15]. Nevertheless, whether as a result of the inherent difficulties in studying a rare disease, or otherwise, this therapy has not been shown to significantly slow decline in lung function or to have a favourable effect on mortality [15], thus additional evidence of its effectiveness and of its mechanism of action are required.
With a prominent presence in a number of airways diseases including cystic fibrosis [16], COPD [17], bronchiectasis and asthma [18, 19], neutrophils are now recognised as central players in the development of persistent chronic inflammation. Studies exploring neutrophil function have reported dysregulated neutrophil chemotaxis in cystic fibrosis [16], COPD [20] and in AATD [4], with increased neutrophil extracellular traps in sputum samples from individuals with bronchiectasis correlating with worse clinical outcomes [21]. However, reports demonstrating high airway neutrophil counts in AATD, even in those with mild lung disease [22], in non-smoking AATD patients [23], and with no evidence of infection or inflammation [23], challenged the hypothesis that altered neutrophil function was affected by a lack of circulating AAT. In concurrence, increased release of primary granule enzymes by neutrophils donated by AATD individuals compared to forced expiratory volume in 1 s (FEV1)-matched non-AATD COPD has been reported, a dysregulation rectified by AAT augmentation therapy [24]. In the current study, we sought to explore the influence of AAT on a further important function of neutrophils, namely reactive oxygen species (ROS) production. Upon activation, oxygen (O2) uptake by neutrophils greatly increases, and transfer of electrons across the plasma or phagosomal membrane by the nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase results in superoxide (O2−) generation [25]. While O2− is only weakly bactericidal, it rapidly dismutates to form H2O2, which can be converted to the strong non-radical oxidant HOCl. An excess of ROS production has been implicated in the development of emphysema and COPD [26] and can arise from neutrophils which are found in high numbers in the airways of individuals with AATD [23]. In patients with COPD who have stopped smoking, phagocytic cells remain a major source of ROS. Oxidative stress can lead to airway epithelial cell injury through a variety of mechanisms including lipid peroxidation, oxidation of proteins and carbohydrates, posttranslational modifications and formation of damage-associated molecular patterns [26]. Oxidative stress can also activate the NF-κB pathway propagating the inflammatory burden in the lung [27]. A range of antioxidant reagents including N-acetylcysteine have been trialled in the treatment of COPD, some with modest success [28]. AAT has been shown, in vitro, to modulate the production of ROS by neutrophils, but mechanisms of inhibition remained unsolved [29]. The current study addresses this knowledge gap and describes the process by which AAT may modulate neutrophil NADPH oxidase activity and ROS production in vitro, and in vivo in response to AAT augmentation therapy.
Materials and methods
Study design
Non-smoking healthy control (HC) volunteers (n=16, 26–45 years), all MM phenotype with serum AAT concentrations in the range of 1.5 g·L−1, were recruited. AATD patients (homozygous for the Z-allele, nonsmokers, n=24) were recruited from the Irish α-1 Antitrypsin Deficiency Registry (FEV1 60.52±3.86% predicted). AATD patients were receiving intravenous AAT augmentation therapy (Zemaira®; CSL Behring, King of Prussia, PA, USA) at 60 mg·kg−1 body weight weekly (n=7). All HC and AATD participants provided written informed consent, approved by the Beaumont Hospital Institutional Review Board (reference 13/92). A Sebia isoelectrofocusing kit was employed for AAT phenotyping with the HYDRASYS system as previously described [30].
Neutrophil isolation and membrane analysis
Blood neutrophils were isolated as previously described [31] and determined 98% pure by flow cytometric analysis employing a monoclonal antibody to CD16b [11, 32]. Neutrophil viability was assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay and found to be >98%. For purification of neutrophil plasma membranes, neutrophils were lysed by N2 cavitation, and a post-nuclear supernatant fractionated by sucrose gradient ultracentrifugation using lysis buffers and centrifugation speeds as previously described [24]. Plasma membrane pellet was prepared for liquid chromatography–mass spectrometry (LC-MS)/MS analysis as outlined in the supplementary methodology and as previously described [24].
Neutrophil activity assays
Oxygen consumption by purified neutrophils was measured in a Clarke-type oxygen electrode as previously described [33]. Neutrophils (2×107 cells in PBS with glucose (PBSG)) reached a steady state of respiration and were then stimulated with interleukin-8 (IL-8) (10 ng/2×107cells) and formylmethionyl-leucyl-phenylalanine (fMLP) (10 µM) in the presence or absence of AAT (27.5 µM; Athens Research and Technology, Inc, Athens, GA, USA).
The production of O2− by neutrophils was measured by cytochrome c reduction assay as described [24], in the presence of increasing concentrations of AAT (1.7–27.5 μM) and remained unstimulated or where exposed to fMLP or fMLP plus IL-8. fMLP is a relevant stimulus to use, as fMLP alone only poorly activates the NADPH oxidase, and pronounced oxidase activity in response to fMLP is observed in primed neutrophils [4]. Thus, fMLP challenge confirmed the purification of un-primed HC cells and, in contrast, the primed state of AATD neutrophils. Absorbance (550 nm) was monitored on a SpectraMax® M3 multi-mode microplate reader (Molecular Devices, San Jose, CA, USA) and O2− concentration determined as previously described [24]. Cyclic AMP (cAMP) was measured using a colorimetric cAMP Assay Kit (Competitive ELISA), according to the manufacturer's instructions (Abcam, Cambridge, UK). The ability of AAT to bind fMLP is as described in the supplementary methodology.
Electrophoresis and immunoblotting
Samples were subjected to SDS-PAGE under denaturing conditions using the Novex® Bis-Tris gel system (Invitrogen™) and transferred to 0.2 µM PVDF Membrane (Roche, Basel, Switzerland) by Western blotting. Immuno-blots were probed with 0.2 µg·mL−1 rabbit polyclonal anti-ERK 1/2 antibody, 1 µg·mL−1 rabbit monoclonal anti-ERK1 (phospho T202), anti-ERK2 (phospho T185) (Abcam), goat polyclonal anti-p47phox, rabbit monoclonal anti-p67phox or rabbit monoclonal anti-HLA (Abcam). Secondary antibodies were horseradish peroxidase (HRP) linked anti-goat or anti-rabbit (Cell Signalling Technology, Danvers, MA, USA). Immunobands were detected using Immobilon™ Western Chemiluminescent HRP-substrate (Millipore, Burlington, MA, USA) solution and a G-Box Chemie XL (Syngene, Cambridge, UK) and analysed using GeneSnap and GeneTools software.
Statistical analysis
Data was analysed using GraphPad Prism version 8 (GraphPad, La Jolla, CA, USA) and presented as mean±sem of biological replicates or independent experiments. One-way or two-way ANOVA was used when comparing three or more groups tested with one or two factors, respectively. A linear mixed-effects model was used when comparing multiple groups of different sizes. A p-value of ≤0.05 was deemed statistically significant following Bonferroni or Tukey post hoc multiple comparison tests. Proteomics results were analysed using Progenesis software. One-way repeated measures (within-subject) ANOVA was used for dependent group comparisons. Differential protein expression was defined as ≥1.5-fold change in expression with a p-value of <0.05.
Results
Exogenous AAT modulates neutrophil oxygen consumption and superoxide anion production in vitro
It has been shown that AAT can modulate O2− production by purified circulating neutrophils in response to non-physiological phorbol ester [29] or serine protease activity [24]. We set out to explore this phenomenon using the more relevant stimuli IL-8 [34] and fMLP [24]. Incubation of purified HC neutrophils with cytochrome c and IL-8/fMLP-was recorded after 5 min (figure 1a). In contrast, AAT significantly lowered levels of extracellular O2− in a dose-dependent manner, with no O2− recorded in the presence of physiological levels of AAT (27.5 µM) (figure 1b). Indeed, significantly lower levels of extracellular O2− were detected following inclusion of 3.4 µM AAT (p<0.0001), and a further reduction in O2− production by cells incubated with 6.8 µM AAT (p<0.0001). However, as methionine residues may act as catalytic antioxidants, and oxidation of methionine 351 or methionine 358 in AAT may influence the detection of O2− [35], from this experiment we could not determine whether AAT was directly influencing the activity of NADPH oxidase or whether AAT was acting as a scavenger of O2−. To this end, the effect of exogenous AAT on cellular O2 uptake was assessed. After addition of IL-8/fMLP to neutrophils, there was a brief lag of about 15 s before O2 consumption commenced, after which it rapidly increased and reached a linear rate up to 240 s (figure 1c). The lag in oxygen consumption increased to about 140 s for neutrophils bathed in exogenous AAT (27.5 µM) (figure 1c). After 240 s incubation, AAT significantly reduced O2 consumption of IL-8/fMLP-stimulated neutrophils from a maximum of 35 nmol to 7 nmol/107 cells (p=0.007) (figure 1d). Collectively, this data demonstrates that AAT may directly inhibit NADPH oxidase activity in response to IL-8/fMLP, and to assess this further, we next evaluated the effect of AAT on downstream signalling pathways related to NADPH oxidase activation.
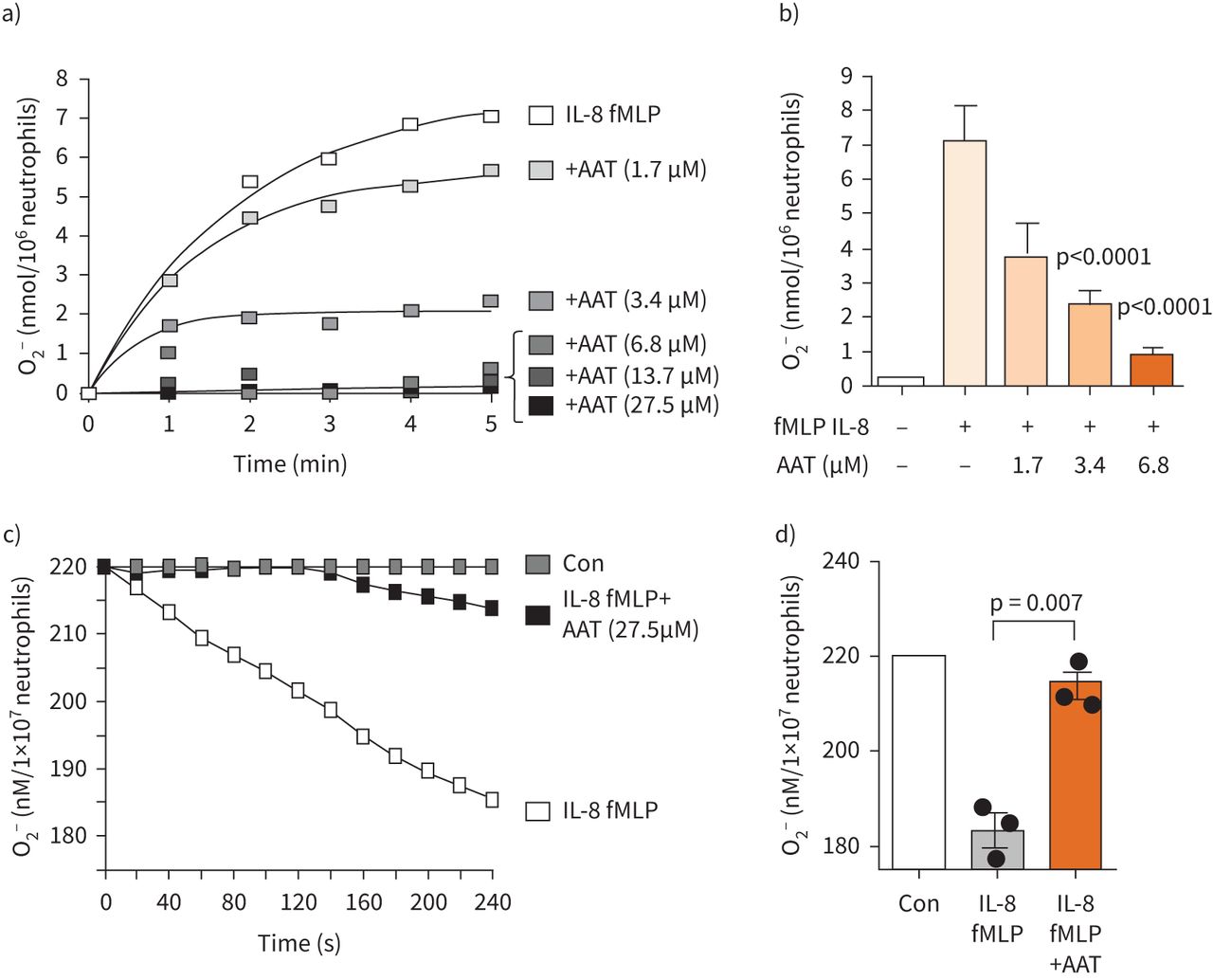
Neutrophil oxygen consumption and superoxide anion production is modulated by α-1 antitrypsin (AAT). a and b) Healthy control (HC) neutrophils (1×107 mL−1) were bathed in AAT (0–27.5 µM) and challenged with interleukin-8 (IL-8) (10 ng/2×107cells) and formylmethionyl-leucyl-phenylalanine (fMLP) (10 μM). O2− production was monitored by cytochrome c reduction assays recorded at 550 nm. b) O2− production was significantly decreased following treatment at 3.4 µM AAT and above (n=3 biological repeats, ANOVA followed by Bonferroni post hoc test). c) Representative profile of continuous monitoring of oxygen consumption by neutrophils exposed to IL-8 (10 ng/2×107cells) and fMLP (10 µM), or in the presence of AAT (27.5 µM). d) HC neutrophils stimulated for 240 s in the presence of AAT (27.5 µM) demonstrated significantly reduced O2 consumption (n=3 biological repeats, Mann–Whitney U-test). Con: control.
cAMP levels and ERK1/2 activation were decreased by exogenous AAT in vitro
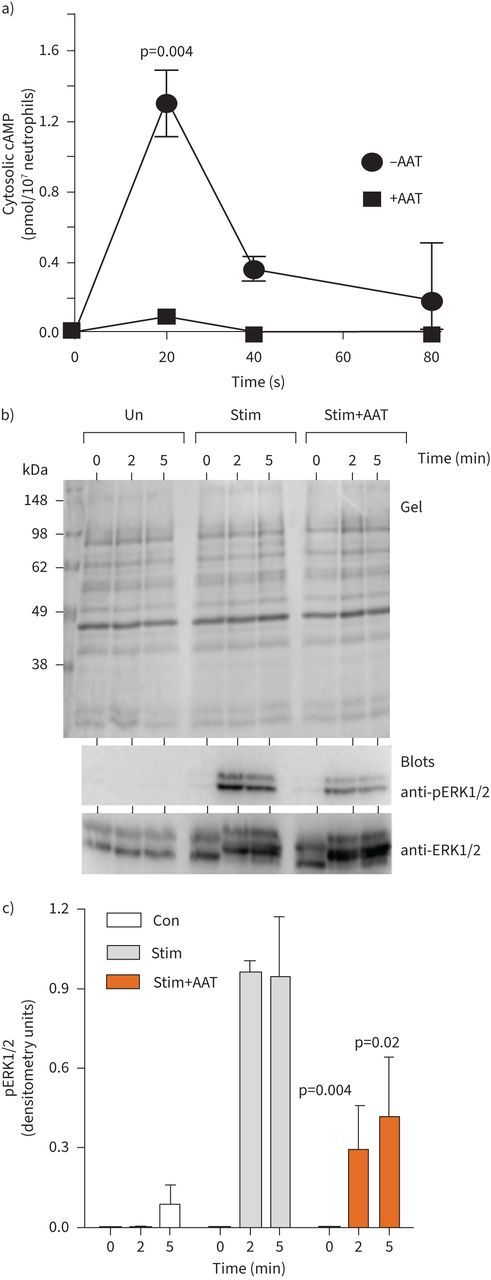
The fMLP receptor is a G-protein-coupled receptor, binding of which leads to dissociation of G-protein subunits and activation of downstream signalling proteins to generate secondary messengers including cAMP, leading to NADPH oxidase activation [36]. Monitoring the intracellular cAMP levels in neutrophils in response to fMLP/IL-8 revealed an increase over the baseline after 20 s (∼1.3 pmol/107 cells, p=0.004), but decreased to the initial level after only 80 s. Moreover, inclusion of exogenous AAT (27.5 µM) significantly reduced levels of cytosolic cAMP in fMLP/IL-8-stimulated neutrophils (p=0.004) (figure 2a).
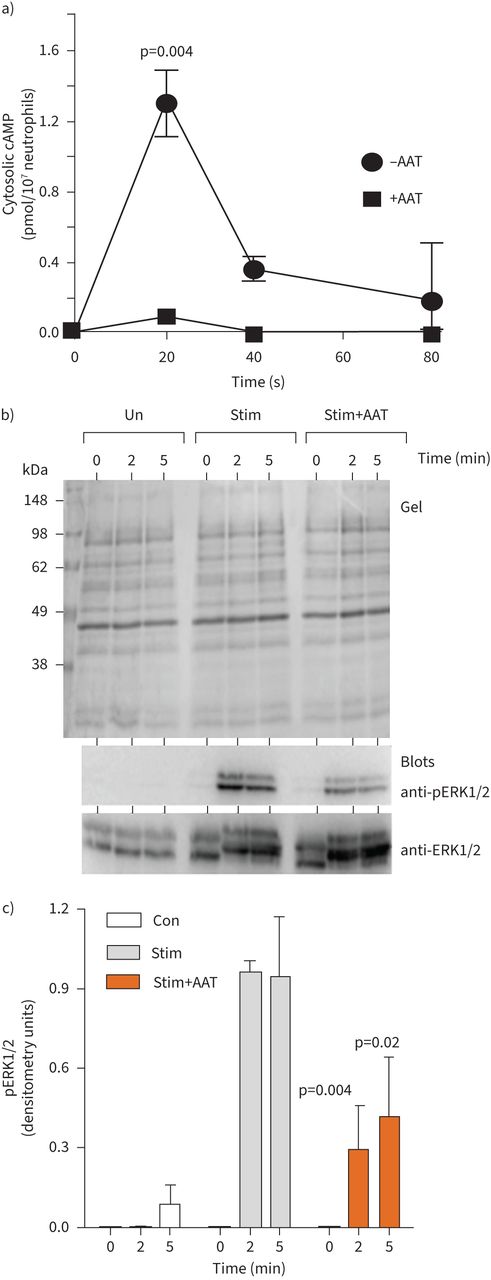
α-1 antitrypsin (AAT) modulates cAMP and kinase activation. a) Neutrophils (1×107·mL−1) were challenged with interleukin-8 (IL-8) (10 ng/2×107cells) and formylmethionyl-leucyl-phenylalanine (fMLP) (10μM) in the presence of α-1 antitrypsin (AAT) (27.5 µM). cAMP levels were monitored by colorimetric competitive ELISA (n=3 biological repeats, p=0.004, two-way ANOVA). b and c) Representative Coomassie blue-stained SDS gel of whole cell lysates of unstimulated neutrophils (Un) or following fMLP/IL-8 stimulation for 60 s or following inclusion of AAT (27.5 µM). Cell fractions were immunoblotted for ERK or pERK. c) pERK levels increased following stimulation (Stim), an effect inhibited by AAT (27.5 µM) at 2 and 5 min (n=3 biological repeats, p=0.004 and p=0.002, respectively, two-way ANOVA).
In response to fMLP, key reactive kinases include activated extracellular signal-regulated kinase (ERK1/2) [37]. We first determined fMLP/IL-8-activated ERK1/2 in isolated neutrophils, with activation monitored by measurement of phosphorylation by immunoblotting using phospho-specific antibodies that recognise the dually phosphorylated motif T202 and T185 within activated ERK1/2. As shown in figure 2b, fMLP/IL-8 triggered phosphorylation of ERK1/2, which was not apparent in unstimulated cell samples. Phosphorylation was detected at 2 min and was maintained after 5 min stimulation. ERK1/2 was also phosphorylated following fMLP/IL-8 activation in the presence of AAT (27.5 µM), but phosphorylation was over three-fold less compared to AAT untreated cells (p=0.004 and p=0.002, at 2 and 5 min, respectively). These results confirm the ability of AAT to modulate key cell signalling events in vitro, and thus the impact on activation of NADPH oxidase components was next explored.
Membrane translocation of p67phox and p47phox in fMLP/IL-8-stimulated neutrophils is inhibited by exogenous AAT in vitro
NADPH oxidase activation involves assembly of integral membrane and cytosolic proteins including p67phox and p47phox. Activation of neutrophils with stimuli such as fMLP/IL-8 promotes the translocation of p67phox and p47phox to the plasma membrane, where they dock with flavocytochrome b558 [38]. Following subcellular fractionation of neutrophils, the plasma membrane fraction is effectively harvested at a 34% (w/w) sucrose interface [39], as depicted in the Coomassie blue-stained gel of figure 3a. Upon fMLP/IL-8 activation for 30 or 60 s, polyclonal antibodies against p67phox and p47phox confirmed a clear shift to plasma membrane fractions. Membranous p67phox and p47phox occurred rapidly and increased 100-fold following fMLP/IL-8 stimulation for 60 s (figure 3b). The effect of exogenous AAT on the translocation and membrane expression of p67phox and p47phox was also determined by immunoblotting of purified plasma membranes. For this analysis, cells were suspended in PBSG containing AAT (27.5 µM) prior to fMLP/IL-8 stimulation. AAT inhibited fMLP/IL-8-stimulated membrane translocation and abundance of p67phox (p=0.002) and p47phox (p=0.005), with levels reduced to baseline (figure 3b).
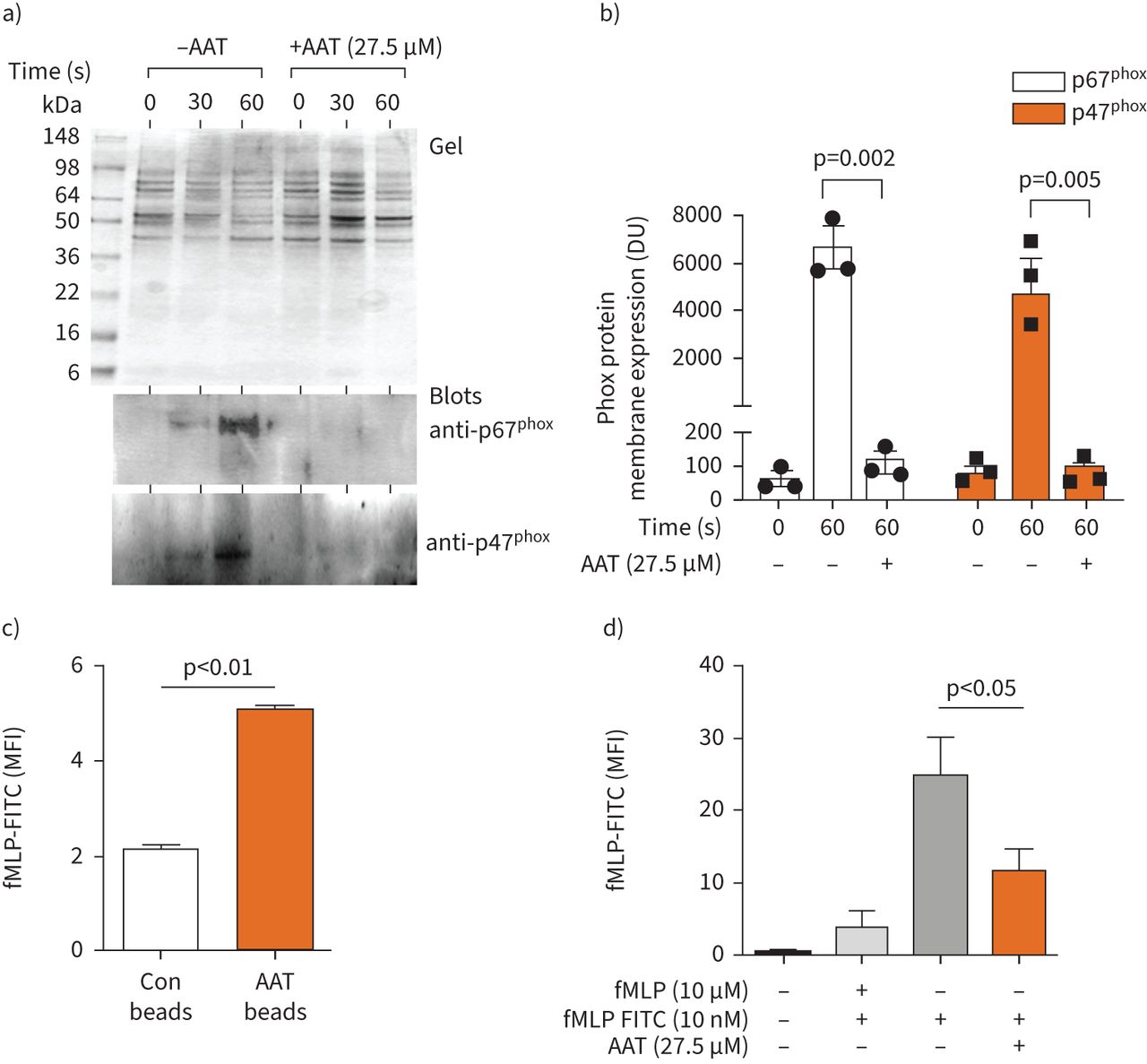
α-1 antitrypsin (AAT) modulates phox protein membrane translocation. a and b) Coomassie blue-stained SDS gel of plasma membranes isolated from unstimulated neutrophils, or following formylmethionyl-leucyl-phenylalanine (fMLP)/interleukin-8 stimulation for 30 or 60 s, in the presence or absence of AAT (27.5 µM) (a; top panel). Membrane fractions were immunoblotted for the presence of p67Phox or p47Phox (bottom 2 panels). b) Translocation of p67Phox and p47Phox to plasma membranes post 60 s stimulation was significantly reduced by AAT (p=0.002 and p=0.005, respectively, two-way ANOVA). Results are expressed as relative densitometry units (DU) of immunoblots. c) Flow cytometry analysis of polystyrene beads either uncoated (control (Con) beads) or coated with AAT and incubated with a fluorescein isothiocyanate (FITC)-labelled fMLP peptide (10 nM). AAT beads bound significantly increased levels of fMLP compared to control beads (n=3, ANOVA, Bonferroni post hoc test). d) Neutrophils were untreated or treated with fMLP or fMLP-FITC in the presence or absence of AAT (27.5 µM) and assessed by flow cytometry. AAT significantly reduced fMLP-FITC neutrophil engagement (n=3, Bonferroni post hoc test). All measurements are mean±sem. MFI: mean fluorescence intensity.
The mechanism by which AAT exerts this inhibitory effect was next explored. We have previously demonstrated that plasma-purified AAT regulates IL-8-induced neutrophil chemotaxis. The mechanism of inhibition involved direct binding of AAT to IL-8, thereby modulating IL-8 engagement with neutrophil membrane CXCR [4, 40]; however, the mechanism by which AAT regulates fMLP signalling remained unsolved. We hypothesised that binding of fMLP to AAT modulated fMLP binding to the specific cell surface receptor N-formyl peptide receptor (FPR). To determine direct binding of AAT to fMLP, experiments were performed with purified AAT protein and fluorescein isothiocyanate (FITC)-labelled fMLP (fMLP-FITC). In initial experiments, AAT-coated polystyrene beads (10 µm) were incubated with fMLP-FITC, and at the same time beads with no AAT functioned as a control. Binding events were analysed by flow cytometry and established significantly enhanced binding of fMLP-FITC to AAT-coated beads (p<0.01) (figure 3c). The ability of fMLP to interact with FPR, in the presence or absence of AAT (27.5 µM), was next explored by flow cytometry. At the dose of 10 nm fMLP-FITC, it is expected that only the FPR will be engaged and stained [41]. In competitive inhibition assays pre-incubation of neutrophils with fMLP caused a significant reduction in fMLP-FITC binding to neutrophil membranes. Similarly, inclusion of AAT in reactions significantly reduced FITC-fMLP neutrophil interaction (p<0.05) (figure 3d).
Collectively, this set of experiments confirms that AAT:fMLP binding prevents fMLP neutrophil membrane engagement, thereby modulating downstream signalling events required for membrane translocation of p67phox and p47phox. Such comparative results prompted an investigation into levels of neutrophil oxidase activity in individuals deficient in AAT.
Increased generation of superoxide anion by circulating neutrophils from patients with AATD in response to fMLP
It has been shown that circulating neutrophils from patients with AATD produce greater levels of O2− than FEV1-matched COPD in response to fMLP or tumour necrosis factor-α (TNF-α)/fMLP [24]. Increased O2− generation by AATD neutrophils in response to fMLP is indicative of priming [38]. We sought to explore priming of neutrophils in AATD, with focus on enhanced O2− production and phox protein membrane expression. As previously described, compared to 1.5 g·L−1 of circulating M-AAT protein in HC samples, AATD patients’ plasma contained ∼0.29 g·L−1, with the Z-AAT protein profile confirmed by isoelectric focusing (figure 4a) [24]. In O2− anion production assays, fMLP (100 nM) weakly activated the NADPH oxidase of neutrophils donated by HC individuals, a result divergent to the effect of fMLP on O2− production by cells of AATD patients (figure 4b). At 5 min there was a 1.5-fold increase in detectable O2− from AATD neutrophils exposed to fMLP (n=11, p=0.002) (figure 4c). Ensuing experiments examined whether the increased NADPH oxidase activity in AATD neutrophils was supported by plasma membrane abundance of components of the NADPH oxidase, p67phox and p47phox. Proteomic analysis of HC neutrophil plasma membranes was carried out and compared to AATD membrane fractions. Differentially expressed proteins included increased abundance of the key NADPH oxidase constituents p67phox (p=0.01) and p47phox (p=0.0004) (figure 4d and e) on AATD plasma membranes. The finding of increased levels of p67phox and p47phox was further confirmed by Western blot analysis. Subcellular membrane fractions were purified and visualised by Coomassie blue staining (figure 4f). By immunoblotting of neutrophil plasma membrane fractions, a 50% increase in the abundance of p67phox and p47phox was detected on AATD membranes compared to HC fractions (p=0.03) (figure 4g). To summarise, these data suggest alterations in O2− production by circulating neutrophils from individuals with AATD compared to HC, supporting increased levels of NADPH oxidase components.

Exaggerated O2− production by primed α-1 antitrypsin (AAT) deficiency (AATD) neutrophils in response to formylmethionyl-leucyl-phenylalanine (fMLP). a) IEF AAT glycan phenotypes. Healthy control (HC) MM AAT glycoforms (M2–M8) and AATD Z-allele glycans (Z2, Z4 and Z6) are presented. Circulating protein levels of M-AAT or Z-AAT AAT (g·L−1) are included. b and c) O2− production by HC or AATD neutrophils following exposure to fMLP (10 µM) or fMLP/interleukin-8 (IL-8) (10 ng/2×107cells) over a 5-min time course. c) In response to fMLP, AATD neutrophils exhibited increased O2− production compared to healthy control neutrophils at 5 min (n=11, p=0.001, t-test). HC and AATD neutrophil plasma membranes were assessed by mass spectrometry. d and e). Increased p67phox and p47phox were detected on AATD plasma membranes compared to HC fractions (n=10, biological repeats, p=0.01 and p=0.0004, respectively, ANOVA, displayed as ArcSinh Normalised Abundance values. f) Coomassie gel of purified neutrophil plasma membrane fractions from HC or AATD individuals. g) Membrane fractions were immunoblotted for p67phox and p47phox, and densitometry of immune-bands performed. Expression of p67phox and p47phox were significantly increased in AATD membrane fractions versus HC samples (n=3 individuals per group, p=0.03, ANOVA Bonferroni post hoc test).
AAT augmentation in vivo reduced neutrophil NADPH oxidase activity in AATD
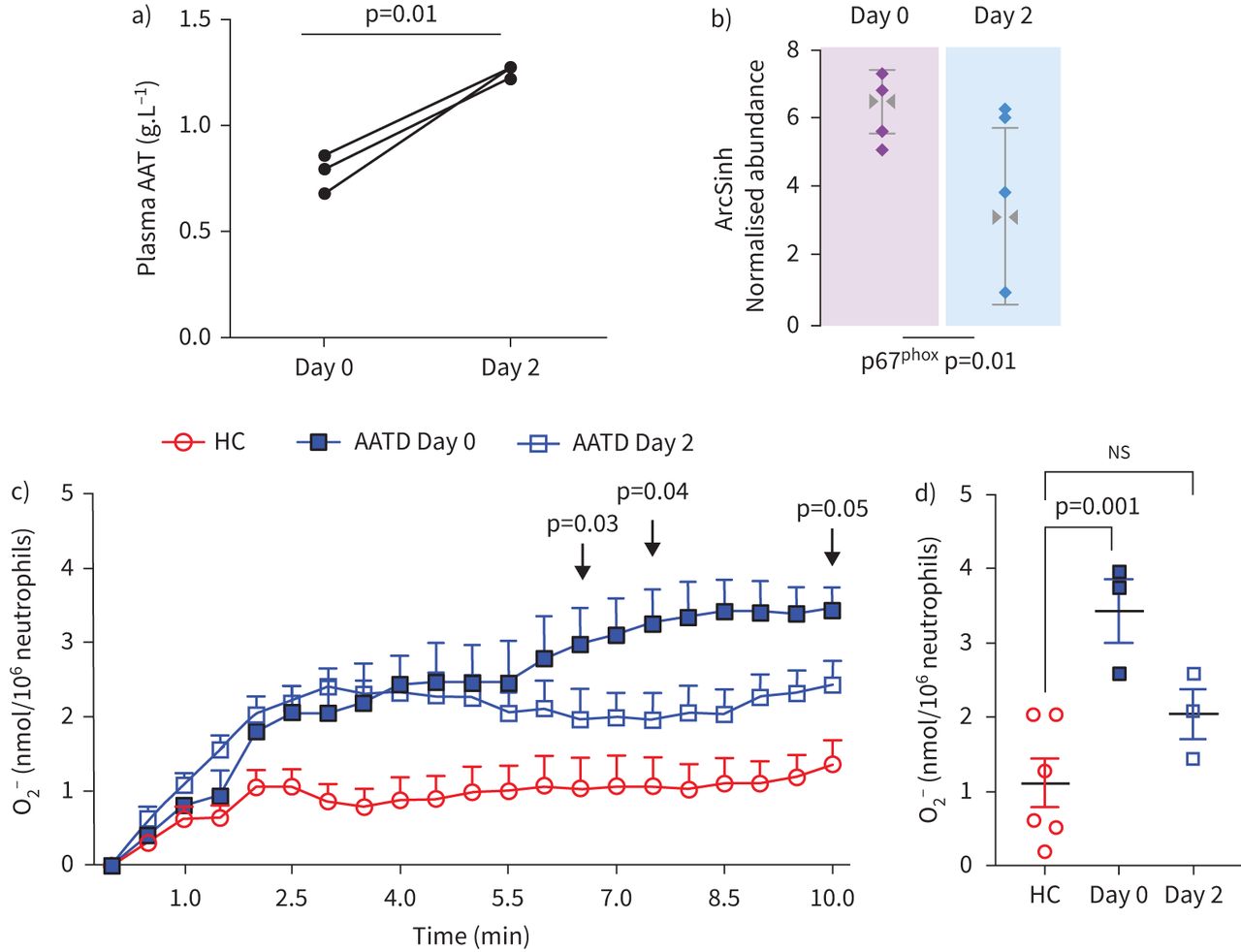
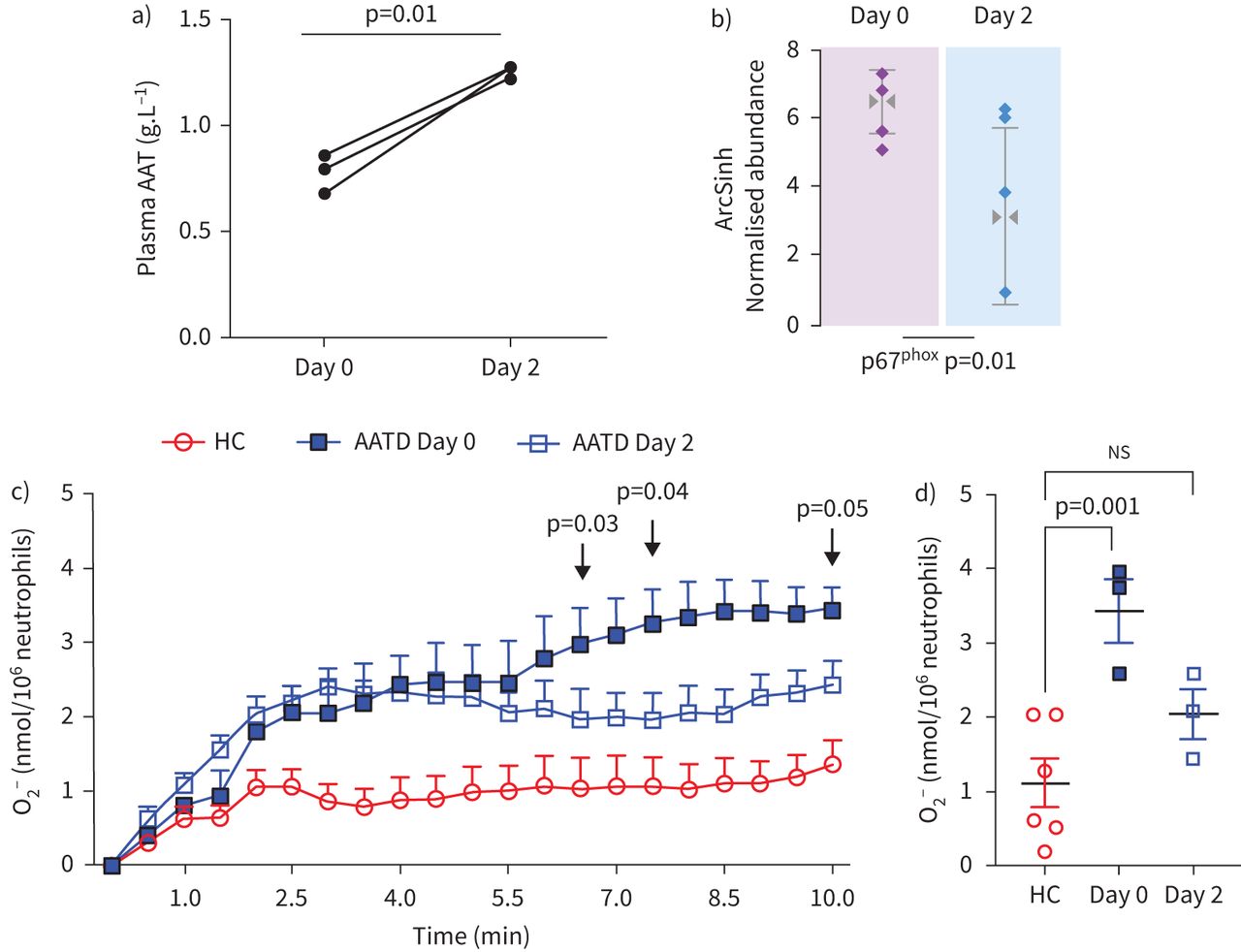
Experiments were carried out to investigate whether AAT augmentation therapy influenced membrane expression of phox proteins in vivo, and in particular, whether the impact of AAT on O2− release by purified AAT in vitro could be validated in vivo post-AAT infusion. For this analysis, patients with AATD donated a blood sample before receiving AAT augmentation therapy (day 0) and a further sample 2 days post therapy (day 2). As previously reported, following AAT therapy, plasma levels of AAT were significantly increased, in comparison to the trough level of prior treatment (∼1.3 g·L−1 and ∼0.8 g·L−1, respectively, p=0.01) (figure 5a) [24]. It has been documented that infused AAT binds to circulating neutrophil plasma membranes and impacts on the neutrophil membrane proteome [24]. By tandem mass spectrometry (MS/MS) analysis no difference in the expression levels of p47phox was observed between day 0 and day 2 of therapy, but in contrast, results revealed p67phox as differentially expressed with significantly reduced levels on day 2 of AAT augmentation therapy compared to day 0 (fold change >1.5, within-subject ANOVA p=0.01) (figure 5b). We next aimed to investigate whether increased plasma levels of AAT, and reduced p67phox membrane expression in response to AAT augmentation therapy, could lead to normalisation of NADPH oxidase activity. Neutrophils were isolated on day 0 and day 2 of AAT augmentation therapy (n=3 paired samples); the characteristics of the burst of O2− production are summarised in figure 5c. AATD neutrophils isolated on day 0 of therapy demonstrated a constant increase in O2− production with 3.43±0.3 nmol/106 cells recorded at 10 min. In contrast, O2− production by AATD neutrophils on day 2 of therapy plateaued at 2.5 min (2.03±0.23 nmol/106 cells), with a significant difference in O2− measured between day 0 and day 2 at 6.5 (p=0.03), 7.5 (p=0.04) and 10 (p=0.05) min stimulation (figure 5c). Moreover, after 10 min fMLP challenge of neutrophils isolated on day 0 of treatment, significantly increased levels of extracellular O2− compared to HC were recorded (3.4±0.3 and 1.2±0.33 nmol/106 cell, respectively) (p=0.001) (figure 5d). In comparative analysis, the level recorded by day 2 augmentation therapy neutrophils, however, was not statistically different to that of HC cells (2.031±0.231 nmol/106 cell), possibly suggesting a shift towards normalisation of NADPH oxidase processes post therapy (figure 5d). Collectively, these results demonstrate that infused AAT therapy in AATD patients leads to reduced assembly of neutrophil NADPH oxidase at the plasma membrane and reduced O2− generation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo intravenous α-1 antitrypsin (AAT) replacement therapy modulates NAPDH oxidase activity of circulating neutrophils in AAT deficiency (AATD). a) Day 0 compared to day 2 plasma levels of AAT post-AAT augmentation therapy (p=0.01, n=3 paired patient samples, paired t-test). b) The abundance of p67phox on plasma membranes of circulating neutrophils was assessed by mass spectrometry analysis. Membrane p67phox was reduced on day 2 post intravenous AAT replacement therapy (n=4 paired patient samples, p=0.01, within-subject ANOVA). c and d) AATD neutrophils (1×107·mL−1) isolated on day 0 and day 2 were challenged with formylmethionyl-leucyl-phenylalanine (fMLP) (10 µM). O2− production by AATD neutrophils on day 0 was significantly higher than levels produced on day 2 post intravenous AAT replacement therapy at 6.5 min (p=0.03), 7.5 min (p=0.04) and 10 min (p=0.05) (n=3, mixed-effects analysis). d) O2− production was significantly increased by day 0 AATD neutrophils compared to healthy control (HC) cells (p=0.001). No significant difference (ns) was recorded between HC neutrophils and day 2 neutrophils (n=3 individuals per group, one-way ANOVA, Tukey's multiple comparisons test at 8.5 min).
Discussion
In this study, we demonstrate that AAT can modulate fMLP/ FPR engagement and downstream signalling events including ERK1/2 activation and translocation of NADPH oxidase cytosolic components to the neutrophil plasma membrane. Moreover, we demonstrate that neutrophils from individuals deficient in AAT are in a primed state, as demonstrated by raised levels of p67phox and p47phox membrane localisation and greater propensity for increased activation of the NADPH oxidase and ROS release. We also show that post-AAT augmentation therapy, increased circulating plasma levels of AAT may function to maintain the circulating neutrophil in a quiescent state, as demonstrated by reduced membrane phox protein localisation and lower levels of superoxide production.
The recognition of AAT as a key inhibitor of serine proteases such as neutrophil elastase led to the theory that emphysema in AATD was due to a lack of AAT [42]. This concept was strongly supported by studies of AATD individuals who demonstrated altered proteolytic activity and corresponding reduction in lung injury in response to AAT augmentation therapy [7]. However, over the last few years it has become clear that the sole purpose of AAT as an antiprotease may not be entirely accurate and does not fully account for the pathophysiology of AATD-related disease. In this respect, susceptibility to conditions characterised by aberrant neutrophilic inflammation, such as granulomatosis with polyangiitis or panniculitis [43], is indicative of abnormal neutrophil function in the absence of standard circulating levels of AAT (27.5 µM). In line with this concept, in a murine model of AATD, AAT augmentation therapy served to protect against emphysema, but oxidised AAT with no antiprotease activity successfully reduced influx of neutrophils into the lungs [44].
Airway oxidative stress is associated with bronchitic symptoms and loss of lung function [45], and thus the impact of AAT on neutrophil ROS production is of particular importance. We have previously demonstrated that unchecked neutrophil elastase (NE) activity can lead to ROS generation via PAR2 activation [24], and that IgG class anti-lactoferrin antibodies in AATD can augment production of ROS [11]. Collectively, these latter studies further implicate neutrophil-derived granule enzymes in the pathophysiology of AATD. However, a prerequisite for neutrophil superoxide anion production is oxygen consumption. In the current study we demonstrate the ability of AAT to reduce superoxide production, but this was most likely not due to the oxidant scavenging capacity of AAT [35], but instead due to blockade of oxygen consumption. Consequently, we demonstrate that downstream cell signalling associated with an increase in intracellular cAMP and ERK1/2 activation were significantly reduced by AAT in vitro. Consistent with these findings, major signalling pathways in early cell activity events are impacted by AAT including inhibition of Akt phosphorylation [4] and activation of the MAPK p38 phosphorylation pathway [46]. Moreover, AAT has been shown to cause a blockade of IκBα degradation, thereby preventing induction of NF-κB-regulated pro-inflammatory mediators [11]. In the current study, the impact of AAT on superoxide anion production was dose dependent and remained significant at 3.4 µM. The dose-dependent response is important to consider. In HC individuals, plasma AAT levels are 20–53 µM, and it is thought that the putative protective threshold is 11 µM [47]. While AAT is found in high abundance in the plasma, its concentration falls as it diffuses to other body compartments. AAT levels of the interstitial fluid, for example, are 10–40 µM and in ELF are 2–5 µM [47]. These compartments are clearly relevant when discussing the development of lung pathologies such as COPD and emphysema. Thus, our finding that 3.4 µM AAT inhibits the superoxide production is clinically relevant, as it suggests that physiological concentrations of AAT in ELF can reduce oxidase activity thus potentially having a protective effect.
NADPH oxidase activity is largely receptor mediated and includes activation or priming via G protein-coupled receptors and cytokine receptors including tumour necrosis factor receptors, toll-like receptors, Fc receptors or integrin receptors. In resting cells, p67phox and p47phox are localised within the cytosol, and upon cell stimulation, GTP-bound Rac2 and the phosphorylated p47phox and p67phox translocate to the membrane to interact with cytochrome b558. Priming, on the other hand, is the phenomenon by which neutrophil challenge by a ligand/stimulus fails to induce O2− but renders neutrophils more responsive to strong activation of NADPH oxidase upon interaction with a further ligand. For example, priming by TNF-α leads to p47phox and p67phox phosphorylation, allowing p67phox to dock with gp91phox at the plasma membrane. Of relevance to this study, it has previously been shown that AAT can regulate the TNF-α signalling axes of neutrophils [11, 12, 48], and thus it is very probable that TNF-α plays a key role in neutrophil priming in AATD. In the current study, we found higher abundance of p67phox and p47phox on neutrophil plasma membranes and confirmed an exaggerated level of superoxide production.
Important work to date has focused on the mechanism by which AAT can inhibit cell processes. For example, the ability of AAT to inhibit apoptosis has focused on cells that are capable of internalising AAT such as airway endothelial cells [49]. Notably, neutrophils have been shown to localise AAT to the outer plasma membrane [4]. This observation, together with our finding of AAT's ability to regulate fMLP-induced NADPH oxidase activity, supports the hypothesis that the mechanism lies at the membrane level in neutrophils. Of note, it is recognised that infused AAT in AATD patients binds circulating neutrophils [24], again localised to outer plasma membranes where it may facilitate AAT's immune-regulatory effects. In the current study AAT was found to bind fMLP, as has previously been shown for IL-8 [4, 40], thereby modulating cognate receptor interaction. Of interest, AAT is post-translationally modified by glycosylation through the addition of N-glycosidically linked oligosaccharides, and during the resolving phase of community-acquired pneumonia (CAP), heavily sialylated forms of AAT are produced [40]. Of major interest, as the symptoms of CAP cleared and C-reactive protein levels decreased, negatively charged sialylated-AAT protein bound IL-8, thereby inhibiting IL-8 CXCR-induced neutrophil chemotaxis. In the current study we did not explore the impact of AAT glycans on fMLP signalling, which is acknowledged as a limitation of the study.
Consistent with in vitro data demonstrating the ability of AAT to modulate NADPH oxidase activity in vitro, in the current study, results revealed that 2 days post-AAT augmentation therapy decreased plasma membrane expression of p67phox, in line with diminished priming of neutrophils in vivo, was recorded. Moreover, AAT therapy reduced the AATD neutrophil NADPH oxidase with a shift towards HC levels, as indicated by comparable levels of superoxide anion production. A limitation to this part of the study is the recruitment of a small number of AATD patients receiving augmentation therapy, and while the data are clear, the sample size means that some caution is needed. Nevertheless, despite this weakness, this study presents confirmation of the effect of infused AAT on phox protein expression and neutrophil membrane NADPH oxidase activity. An additional issue to discuss is that the influence of AAT was evident at the peak concentration of AAT therapy on day 2, but by day 0, when plasma AAT levels troughed, circulating neutrophils again exhibit increased oxidase activity. This suggests the possible need for increased AAT dosing as has recently been suggested [50], or alternatively, therapies for AATD patients that may function to avoid the peak and trough cycles to maintain a constant protective AAT level. A further limitation to discuss is the cell isolation method employed which isolates granulocytes and yielded a neutrophil population of 98%. The remaining 2% of cells may include eosinophils. Although eosinophils respond to fMLP [51], it is unlikely that they contributed to the end-point of this study, as it has been reported that eosinophils do not express IL-8 receptors CXCR1 and CXCR2 [52] and FPR1 expression on circulating human eosinophils is very low compared to blood neutrophils of the same subject [53].
The recognition of the role of ROS in the pathogenesis of emphysema in genetic and non-genetic COPD is growing. Our study identifies increased neutrophil NADPH oxidase and superoxide anion production by neutrophils of patients with AATD. Furthermore, it describes a mechanism whereby AAT modulates neutrophil signalling by binding fMLP thereby preventing receptor engagement. In patients receiving AAT augmentation, phox protein membrane assembly and oxidase activity was reduced after treatment. This study supports the use of AAT augmentation therapy to normalise neutrophil ROS production in AATD.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00234-2021.SUPPLEMENT
Supplementary material HAWKINS SUPPLEMENTARY MATERIALS AND METHODS
Acknowledgements
We sincerely thank the Alpha-1 Foundation Ireland, individuals with α1-antitrypsin deficiency and healthy donors for their participation in this study.
Footnotes
Provenance: Submitted article, peer reviewed.
This article has supplementary material available from openres.erjournals.com
Author contributions: P. Hawkins, T. McEnery, D.A. Bergin, N.G. McElvaney and E.P. Reeves contributed to study design. P. Hawkins, T. McEnery, C. Gabillard-Lefort, D.A. Bergin, B. Alfawaz, V. Shutchaidat, P. Meleady, M. Henry, O. Coleman and M. Murphy performed experiments, and analysed and interpreted the data. P. Hawkins, N.G. McElvaney and E.P. Reeves wrote the manuscript.
Conflict of interest: N.G. McElvaney reports support for the present manuscript from the US Alpha One Foundation. E.P. Reeves reports support for the present manuscript from the Medical Research Charities Group/Health Research Board Joint Funding Scheme (MRCG-2018-04). The remaining authors have nothing to disclose.
Support statement: In support of this work, E.P. Reeves acknowledges funding from the Medical Research Charities Group/Health Research Board Joint Funding Scheme (MRCG-2018-04). N.G. McElvaney acknowledges funding from the US Alpha-1 Foundation. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 6, 2021.
- Accepted August 29, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










