





Abstract
Rheumatoid arthritis-associated interstitial lung disease (RA-ILD) is relevant for the prognosis in patients with RA. Nintedanib, which inhibits both receptor and non-receptor type tyrosine kinases, is an antifibrotic drug for the treatment of progressive fibrosing ILDs, such as idiopathic pulmonary fibrosis and systemic sclerosis-associated interstitial lung disease. Little is known about the effects of nintedanib on RA-ILD. We examined the characteristics of a novel induced RA-ILD (iRA-ILD) mouse model and the effects of nintedanib on the model.
D1CC×D1BC mice are highly susceptible to arthritogenic antigens, such as bovine type II collagen, resulting in severe inflammatory arthritis. ILD develops after joint inflammation is alleviated. Serum surfactant protein D levels were monitored as an ILD marker. Nintedanib was orally administered to iRA-ILD mice for 2 months.
The iRA-ILD model showed similar symptoms to those in patients with RA-ILD. The histopathological features of pulmonary disorder resembled nonspecific interstitial pneumonia, but with metaplastic epithelium. Histopathological analysis revealed that in addition to reducing fibrosis, nintedanib suppressed M2 macrophage polarisation and hyperplasia of Type 2 alveolar epithelial cells. The metaplastic epithelium acquired invasiveness because of the expression of E-cadherin, MMP7, Tgf-β, Col1a1, Padi2 and Padi4. Moreover, citrullinated peptides were detected in these invasive epithelial cells as well as in the bronchiolar epithelium. Administration of nintedanib reduced the expression of Pad4 and citrullinated peptides and eliminated invasive epithelial cells.
The broad inhibitory effects of nintedanib on tyrosine kinases may contribute to the overall improvement in RA-ILD, including epithelial abnormalities associated with progressive lung fibrosis.
Abstract
A novel induced RA-ILD mouse model develops progressive lung fibrosis similar to NSIP, with epithelial abnormalities. Nintedanib provides a therapeutic intervention in the model, suggesting evidence of improvement in patients with RA-ILD. https://bit.ly/3yivzt1
Introduction
Rheumatoid arthritis (RA) is caused by chronic inflammation of the joints. RA-associated interstitial lung disease (RA-ILD) is a serious extra-articular manifestation of RA. The current prevalence of RA-ILD in patients with RA is ∼20–50% [1]. In addition, drug-induced lung disease by disease-modifying anti-rheumatic drugs, such as methotrexate and leflunomide, presumably affects the onset of RA-ILD. Most RA-ILD is classified as nonspecific interstitial pneumonia (NSIP) and usual interstitial pneumonia (UIP) patterns [2]. The mortality rate of RA-ILD is increasing, even though treatment with biologics has reduced the symptoms of arthritis [3].
The developmental mechanism of RA-ILD is not clear, one of the reasons being the lack of appropriate animal models [4]. For example, in a collagen-induced mouse model, there are no symptoms related to lung disease. SKG mouse is known as a mouse model of RA-ILD, in which infiltration of lymphoid cells and fibrosis develops. The onset of its symptoms is observed in ∼15% of arthritogenic antigen-treated mice [5]. We previously reported a new murine RA-ILD model, D1CC and D1BC RA mouse model, which expresses human class II transactivator, CIITA, and murine B7.1 in chondrocytes, respectively [6, 7]. Both mice models developed chronic inflammatory arthritis with low dose of bovine Type II collagen (bColII) immunisation. After remission of joint inflammation, we observed ILD without any additional stimuli. Histopathological features in D1CC and D1BC mice are similar to NSIP due to observation of infiltrated lymphoid cells and fibrosis. H2-treatment improved fibrosis in D1CC mice as an RA-ILD model [8]. In this study, we used D1CC×D1BC transgenic (tg) mice (hereafter named iRA-ILD mouse). D1CC×D1BC tg mice are also susceptible to inhalation injury with bleomycin in the lung. It causes chronic interstitial pneumonia, a pathological feature of honeycombing [9].
Serological features in patients with RA are associated with high levels of serum rheumatoid factor (RF) IgG, IgM and anti-citrullinated protein/peptide antibody (ACPA), which are risk factors for extra-articular manifestations, including ILD [4]. Combined presence of ACPA and RF leads to severe systemic inflammation and rapid exacerbation of RA [10]. Both serum RF IgG and IgM levels are strongly associated with the development of RA and RA-ILD [11]. These factors may be involved as a shared disease mechanism leading to the development of RA and ILD [2].
Epithelial–mesenchymal transition (EMT) is an active, reversible process associated with embryogenesis, wound healing, cancer metastasis and fibrosis [12]. Aberrant deposition and remodelling of the extracellular matrix (ECM) have been observed in patients with RA-ILD. Metastatic respiratory epithelia have been observed in the lungs of patients with IPF [13]. The involvement of these ectopic and hyperplastic cells in fibrosis is not clear, but some of them acquire the ability of EMT [14]. Fibrosis and remodelling failures occur as a result of chronic parenchymal damage. Indeed, hyperplastic respiratory epithelial cells proliferate and migrate in the lung [15]. Recent studies have demonstrated that inhibition or silencing of peptidylarginine deiminase 4 (PAD4) reduces EMT and neutrophil extracellular traps [16]; however, the effect of PAD4 on EMT in cancer cells is controversial [17]. Nevertheless, various tumours, including lung squamous carcinoma, express PAD4 and citrullinated peptide, and their function in epithelial cells is less clear [18, 19]. As PAD2 and PAD4 are also involved in EMT, citrullination by PADs may result in tumorigenesis [20].
Nintedanib (BIBF-1120), a drug for patients with progressive fibrosing ILD [21], blocks the ATP-binding pocket of non-receptor tyrosine kinases, such as Src family kinases as well as receptor tyrosine kinases, such as platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR) and vascular endothelial growth factor receptor (VEGFR) [22–24]. Even though pulmonary fibrosis in SKG mice as an RA-ILD model has a lower incidence (∼20%), nintedanib reduced it [25]. Nintedanib inhibited transforming growth factor-β (TGF-β) signalling and downregulated ECM proteins such as Type I collagen [26, 27]. Nintedanib also inhibited macrophage activation and reduced the number of M2 macrophages in the model of systemic sclerosis [28].
Nintedanib has shown broad-spectrum activity against receptor and non-receptor tyrosine kinases and blocks various signalling cascades. Thus, in addition to direct inhibition of lung fibroblasts, it is expected to have multiple effects on other tissues in the lung. In this study, we investigated whether treatment with nintedanib resulted in improvement of histopathological symptoms in iRA-ILD mice.
Materials and methods
Animal care and use
D1CC×D1BC mice were produced by mating D1CC and D1BC transgenic mice and breeding in a pathogen-free animal care facility of Nagoya City University Medical School, according to the guidelines of the facility.
Induction of inflammatory arthritis and nintedanib administration
Inflammatory polyarthritis was induced as previously described [6, 7]. Briefly, mice were anaesthetised with isoflurane and immunised with bColII (0.01 mg/mouse) with an equal volume of complete (1st) and incomplete (2nd–5th) Freund's adjuvant. The first immunisation was given 8 to 10 weeks after birth. The mice were monitored by scoring the joints. Nintedanib was dissolved in 0.5% methylcellulose and administered daily (90 mg·kg−1) by oral gavage from weeks 35 to 43 after the first immunisation (figure 1a). Serum samples were collected from the jugular vein at week 43. The left lobe of the lung was used for histopathological analysis, and the right lobe was used for Western blotting.

Nintedanib attenuates development of interstitial lung disease (ILD) after inflammatory arthritis. a) The schematic diagram shows the time course of bovine Type II collagen (bColII) immunization and the oral administration of nintedanib, which is done daily for 2 months from 35 weeks after first immunization. Arrows indicate bColII immunization (black: bColII/complete Freund's adjuvant, grey: bColII/incomplete Freund's adjuvant). b) The incidence of ILD is determined by overall cut-off of serum SP-D (>53.9 ng·mL−1) at 43 weeks after the first immunization of bColII of nine mice in each group. c) Serum SP-D levels are measured at 43 weeks after first immunization of bColII. Data are presented as mean±SE of nine mice for each group (a–c). Each symbol represents vehicle (white circle), nintedanib (black circle) and age-matched control (black triangle), respectively. d) and e) Histopathology by haematoxylin–eosin (H&E) staining (d) and triple immunohistochemical staining for podoplanin (Pdpn, red), E-cadherin (Ecad, green) and SP-C (white) are performed in vehicle, nintedanib-treated or age-matched control mice (e). Scale bars=50 μm (black and white). f–h) The expression of SP-A (f), -C (g) and -D (h) in the lung tissue is assessed by Western blotting. Data are presented as mean±se of four mice for each group. *p<0.05; **p<0.001, compared with vehicle group. n.s., nonsignificant.
ELISA
Quantitation of serum SP-D (Yamasa, Chiba, Japan) and RF IgG and IgM (Fujifilm Wako Shibayagi, Gunma, Japan) concentrations was determined using ELISA according to the manufacturer's instructions. DBA, D1CC and D1BC mice were used as controls. The cut-off value of serum SP-D was 53.9 ng·mL−1 and was evaluated in a laboratory study [8].
Multiplex cytokine assay
The Bio-Plex Mouse Cytokine 8-Plex (Bio-Rad) was used to quantify the concentration of serum cytokines (interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-10, granulocyte–macrophage colony-stimulating factor (GM-CSF), interferon-γ (IFN-γ) and tumour necrosis factor-α (TNF-α)). The assay was performed according to the manufacturer's instructions.
Serum AST and ALT
Serum AST and ALT levels were quantified using the transaminase Cii test Wako (Fujifilm) according to the manufacturer's instructions.
Histopathology and immunohistochemistry
The mice were euthanised and the lungs harvested and fixed with 4% paraformaldehyde diluted in PBS. Paraffin sections (2 μm) were stained with haematoxylin–eosin (H&E), Masson's trichrome and the following primary antibodies for immunohistochemistry: rabbit anti-E-cadherin, rabbit anti-α SMA, rabbit anti-vimentin, rabbit anti-MMP7 (Cell Signalling Technology, Danvers, MA, USA), rabbit anti-SP-C (Hycult Biotech, Uden, Netherlands), rat anti-Podoplanin (MBL, Aichi, Japan), rabbit anti-collagen I (Novus Biologicals, Littleton, CO, USA), rat anti-F4/80 (Bio-Rad Laboratories, Hercules, CA, USA), rabbit anti-CD3 (Genemed Biotechnologies, San Franciso, CA, USA), rat anti-PTPRC/CD45R (Aviva Systems Biology, San Diego, CA, USA), rabbit anti-arginase I (GeneTex, Irvine, CA, USA), mouse anti-peptidyl-citrulline (F95) and rabbit anti-S100A4 (Millipore, Burlington, MA, USA) and mouse anti-Laminin γ2 N-terminal fragment (γ2pf; Funakoshi, Tokyo, Japan). Histofine Simple Stain Mouse MAX-PO secondary antibodies (Nichirei, Tokyo, Japan) and the Opal 4-colour Fluorescent IHC kit (PerkinElmer, Waltham, MA, USA) were used according to the manufacturer's protocol. All images were captured by fluorescence microscopy (BZ-X710, Keyence) and analysed by hybrid cell count (Keyence, Osaka, Japan) or Fiji.
Western blotting
Western blotting analysis was performed using the ECL system. The following primary antibodies were used: rabbit anti-PDGFRα, rabbit anti-PDGFRβ (Cell Signalling Technology), rabbit anti-Pad4 (MBL), rabbit anti-SP-C (Hycult Biotech), goat anti-SP-D (R&D Systems, Minneapolis, MN, USA), rabbit anti-COL1A1 (Boster Biological Technology, Pleasanton, CA, USA), rabbit anti-SP-A and mouse anti-tubulin-α (Santa Cruz Biotechnology, Dallas, TX, USA) antibodies. Statistical analysis of the expression levels of each protein was performed using Fiji.
In situ hybridisation
In situ hybridisation for Scgb1a1, Sftpc, Col1a1, Tgf-β, Krt5, Krt17, Padi2 and Padi4 was performed using the RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics, Newark, CA, USA), according to the manufacturer's instructions.
Statistical analyses
The results are shown as mean±SE. The significance of differences between vehicle, nintedanib and age-matched control was calculated by one-way analysis of variance followed by Dunnett's test for multiple comparisons. t-test was used to analyse the AST and ALT levels in the serum, as shown in supplementary Figure 2. A value of p<0.05 was considered statistically significant.

Nintedanib attenuates lung fibrosis. a) Masson's trichrome staining for vehicle, nintedanib-administrated or age-matched lung specimen. b) All images for the section from left lung and accessory lobe are captured at a magnification of 200×, and blue fibrotic areas for the total lung areas are calculated by Fiji. Data are presented as mean±SE of nine mice for each group. c) Immunohistochemical staining for Type I collagen (Col1, green), vimentin (red) and αSMA (white). Scale bars=100 μm (white and black). d) and e) Type I collagen (d) and αSMA (e) expressing areas in left lung and accessory lobe are calculated by Fiji. The calculation of stained images is done in the same way as in (b). Data are presented as mean±SE of five mice for each group. f–h) Type I collagen (f), PDGFRα (g) and -Rβ (h) in the whole lung tissues are assessed by Western blotting. Data are presented as mean±SE of four mice for each group. *p<0.05 compared with vehicle group. n.s.: nonsignificant.
Results
D1CC×D1BC mouse has high susceptibility to RA-ILD
Approximately 96% of D1CC×D1BC mice underwent severe joint inflammation after first immunisation with low-dose bColII (supplementary Figure 1a). Although most D1CC+/− mice showed chronic and slow disease progression [6], 70% of D1CC×D1BC mice showed acute severe swelling and redness of the entire paw from the initial onset (supplementary Table 1).
Serum concentrations of RF IgG and IgM in D1CC×D1BC, D1CC, D1BC mice and their genetic background DBA/1J were measured as controls. Serum RF levels in D1CC×D1BC mice were higher than those in single transgenic or DBA/1J mice, even in the absence of stimulation with the arthritogenic antigen (supplementary Table 2). High serum RF levels suggest that D1CC×D1BC mice are more susceptible to joint inflammation than single transgenic mice. In contrast, serum SP-D as an ILD biomarker did not differ. The onset of ILD was determined by increased serum SP-D levels (>53.9 ng·mL−1) from ∼35 weeks after bColII immunisation (supplementary Table 1 and supplementary Figure 1b). Therefore, this increase in serum SP-D indicates the progression of lung fibrosis in D1CC×D1BC mice [8, 21].
Nintedanib reduced the incidence of ILD
We examined whether nintedanib attenuated arthritis-associated ILD. The oral administration of nintedanib or vehicle was started from 35 weeks after the first immunisation and continued daily for 2 months (figure 1a). The incidence of interstitial pneumonia (IP) determined by serum SP-D above the cut-off was ∼65% in bColII immunised D1CC×D1BC mice (hereafter, this induced-RA-ILD mouse model is referred to as iRA-ILD) (figure 1b). Administration of nintedanib decreased the incidence of IP (figure 1b). However, this effect was not reflected in the mean serum concentration of SP-D (figure 1c). The disease progression in iRA-ILD mice was slower than expected, and no deaths occurred during this study. Mortality was the same for both vehicle- and nintedanib-treated iRA-ILD mice (supplementary Figure 2a). No adverse liver effects were observed in D1CC×D1BC mice treated with nintedanib (supplementary Figure 2b–d).
Next, we studied histopathological features using antibodies against E-cadherin (epithelial bronchiole), podoplanin (type I alveolar epithelial cells, AEC1) and SP-C (type 2 alveolar epithelial cells, AEC2). Metaplastic bronchioles, hyperplastic AEC2 and punctured podoplanin-positive areas were observed in the vehicle; however, these features were remodelled by nintedanib treatment (figure 1d and e). We also analysed the expression of surfactant proteins A, C and D in the whole lung by Western blotting. Administration of nintedanib reduced the protein levels of these surfactant proteins (figure 1f–h).
Nintedanib suppressed fibrosis and ColI accumulation
First, to confirm whether nintedanib administration suppressed fibrosis, Masson's trichrome-positive areas (blue-stained regions) were analysed by ImageJ as a fibrotic area. Nintedanib reduced the proportion of fibrotic area (figure 2a and b). For further analysis of ECM deposition, Type I collagen (Col1)- and αSMA-positive areas were calculated from immunohistochemical staining, showing a reduction in both positive areas (figure 2c–e). The expression level of Type I collagen in the whole lung was restored by nintedanib (figure 2f). As nintedanib inhibits the signalling cascade of PDGFR, we also examined whether nintedanib alters the expression levels of PDGFRα and PDGFRβ in the lung. The expression level did not vary with nintedanib administration (figure 2g and h). Therefore, administration of nintedanib attenuates pulmonary fibrosis associated with ECM deposition in iRA-ILD mice.
The number of M2 macrophages was decreased by nintedanib
As the histopathological features of iRA-ILD mice represent NSIP-like pathological features, we studied infiltrated lymphocytes in the lung. Alveolar macrophages tended to polarise M2 rather than M1 phenotype in pulmonary fibrosis [29]. The number of infiltrated macrophages (F4/80), arginase I and F4/80 positive M2 macrophages, B-cells (CD45R) and T-cells (CD3) was counted in the lung by immunohistochemical staining (figure 3). Only the number of M2 macrophages was decreased by nintedanib, suggesting that the effect of nintedanib is associated with polarisation from M2- to M1-macrophage (figure 3c–f). Next, we compared serum cytokine levels in nintedanib- or vehicle-treated iRA-ILD mice. Most M2 macrophage-related serum cytokines, such as IL-4, IL-5 and IL-10, were slightly reduced. Levels of M1 macrophage-related cytokines other than IFN-γ were similar or slightly decreased (supplementary Figure 3).
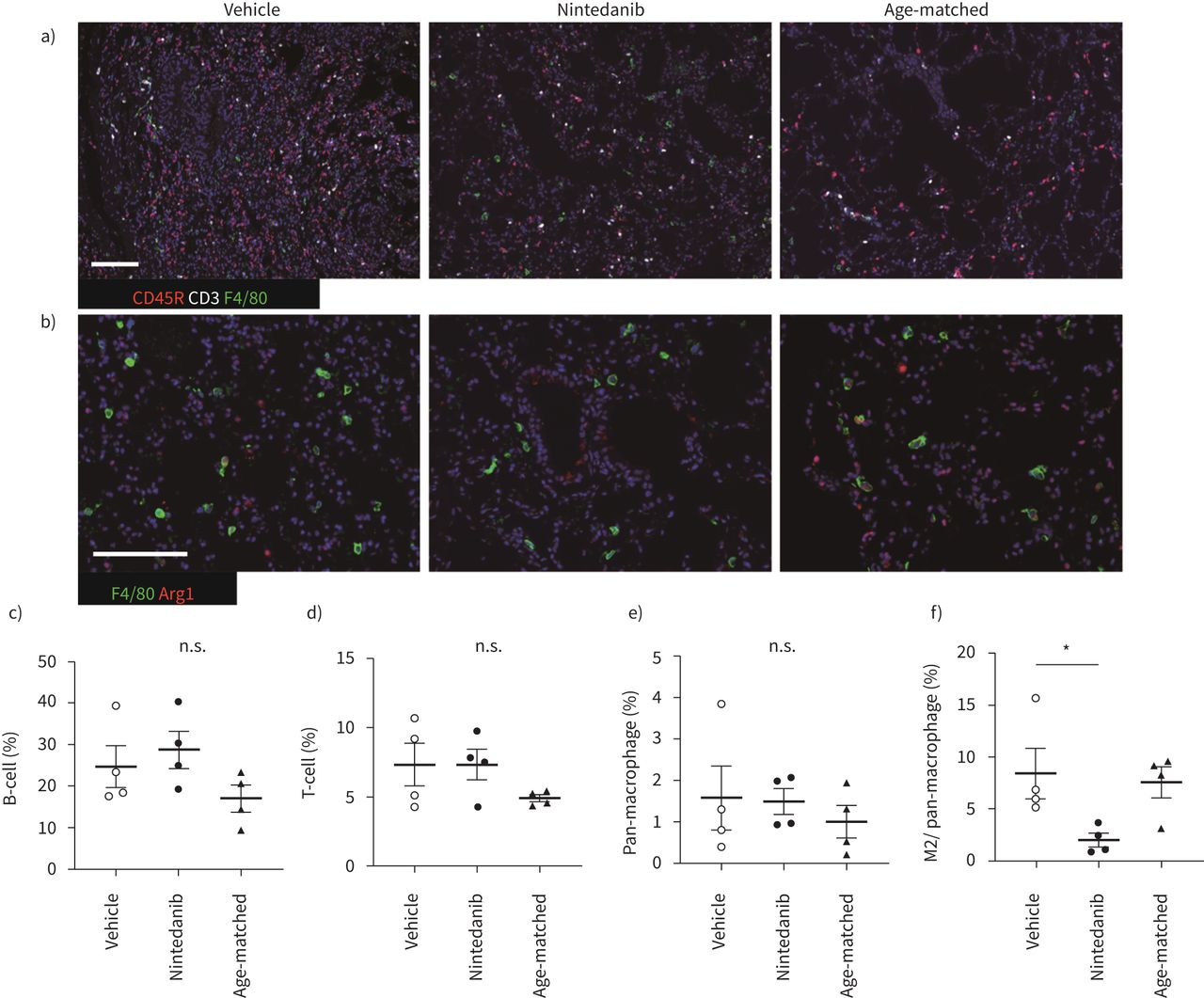
M2 macrophage polarisation was suppressed by nintedanib treatment. a and b) Immunohistochemical staining for F4/80 (green), CD45R (red) and CD3 (white) (a), F4/80 (green) and arginase I (red) (b). Scale bars=50 μm. c–f) The percentage of each infiltrated lymphoid cell is calculated. B-cells (c), T-cells (d), pan-macrophages (e) and the ratio of M2 macrophage in pan-macrophages (f). Images from each immunohistochemical staining in left lung and accessory lobe, which are captured at a magnification of 100× and calculated the ratio of each lymphoid cell for total nuclear cells by Fiji or M2 macrophages in pan-macrophages by hybrid cell count. All data are presented as mean±se of four mice in each group. *p<0.05 compared with vehicle group. n.s.: non-significant.
Nintedanib suppressed ILD-related epithelial abnormalities
To confirm the histopathological characterisation of metaplastic epithelial cells (figure 1e), we stained these cells with antibodies against E-cadherin (epithelial cells) and EMT-related marker, S100A4. E-cadherin and S100A4 double-positive cells were a minor population. E-cadherin single-positive epithelial cells, E-cadherin, and S100A4 double-positive cells were eliminated by nintedanib (figure 4a). Immunohistochemical staining with antibodies against E-cadherin, γ2pf and MMP7 was performed to determine whether metaplastic cells acquired invasiveness. E-cadherin positive cells, including metaplastic cells, expressed MMP7; however, most metaplastic cells also expressed γ2pf simultaneously (figure 4b). We concluded that these metaplastic cells migrate from respiratory bronchioles along with degradation of laminin at the basement membrane by metalloproteases such as MMP7 (hereafter, these metastatic cells named invasive epithelial cells) [30]. Thus, nintedanib suppressed the development of invasive epithelial cells. In situ hybridisation was also performed for the expression of Sftpc, Scgb1a1 and a basal cell marker, Krt5 (supplementary Figure 4). Scgb1a1-positive invasive epithelial cells were distinguished from Sftpc-positive hyperplastic ACE2s (figure 4c). Most Scgb1a1-positive invasive epithelial cells and Sftpc-positive hyperplastic ACE2s vanished with nintedanib treatment, but no overt effects for Sftpc- and Scgb1a1-positive bronchioalveolar stem cells (in the square of the nintedanib-treated specimen, figure 4c).

Nintedanib suppressed the development of invasive epithelial cells and hyperplastic AEC2. a) Immunohistochemical analysis using E-cadherin (Ecad, green) and S100A4 (red). S100A4 single positive cells are estimated as fibroblasts and lymphoid cells including T-cells and macrophages. b) Immunohistochemical analysis using E-cadherin (Ecad, green), γ2pf (red) and MMP7 (white). Invasive epithelial cells secrete MMP7 and degrade laminin (inset a). The administration of nintedanib suppressed the occurrence of these invasive epithelial cells (inset b). c) In situ hybridisation is performed using Scgb1a1 (green) and Sftpc (red). Most Scgb1a1-positive invasive epithelium do not express Sftpc simultaneously. No overt effect by nintedanib on a limited number of Scgb1a1 and Sftpc-positive BASCs is observed (square in vehicle). Scale bars=50 μm (white), 20 μm (yellow) and 10 μm (cyan).
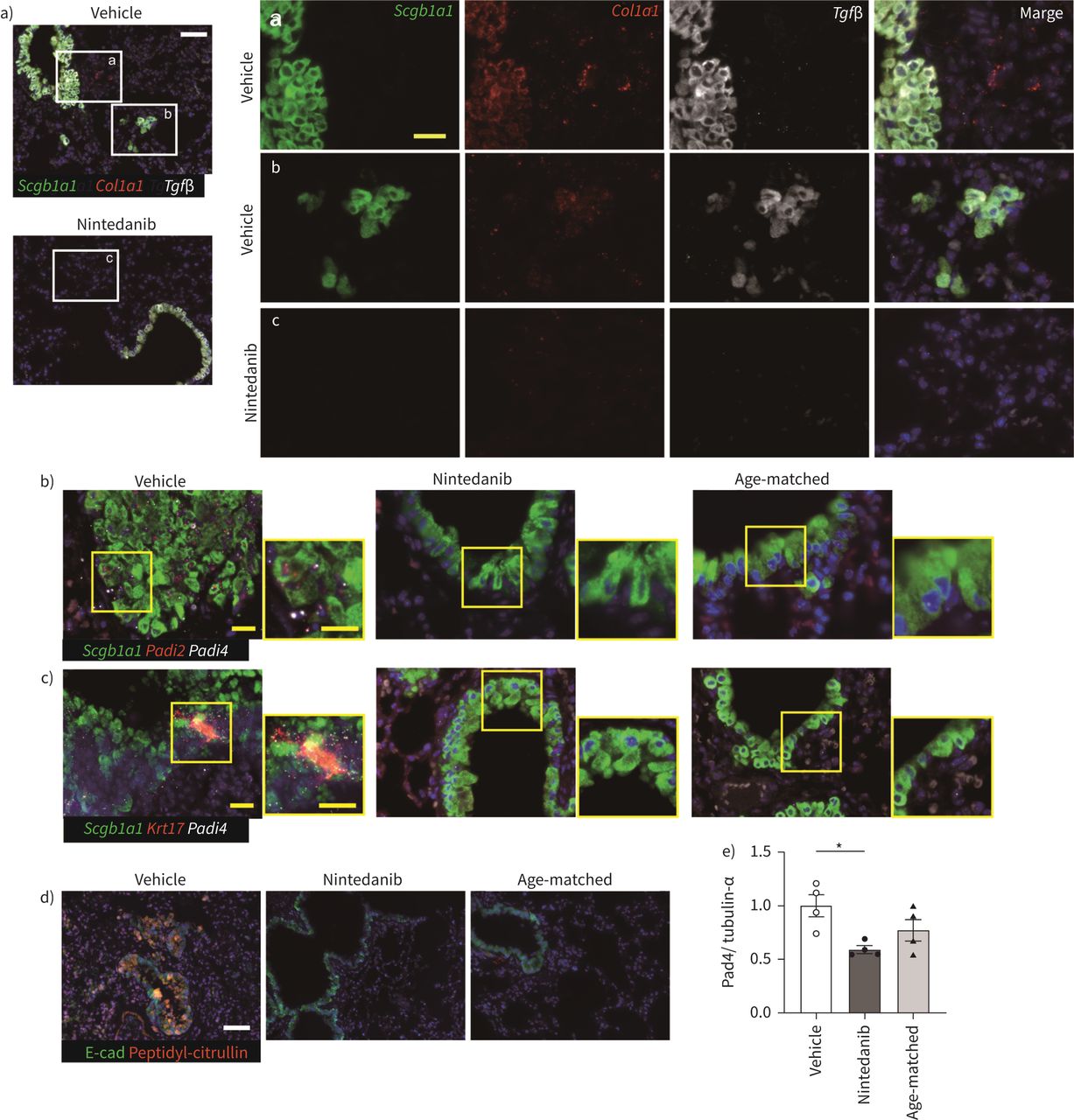
Expression of Padi4 and Padi2 was reduced in epithelial cells by nintedanib
TGF-β is expressed in alveolar macrophages and epithelial cells in IPF. A recent study of single cell analysis also demonstrated that Tgf-β and collagen are expressed in epithelial cells [31]. We tested the expression of Tgf-β and Collagen1a1 (Col1a1) along with Scgb1a1 in the lung. Both signals were detected in respiratory bronchioles and invasive epithelial cells in iRA-ILD mice (figure 5a and Supplemental Figure 5). The expression of Padi4 in these respiratory epithelia was also examined. As autoantibodies such as ACPA are associated with the clinical evidence of RA-ILD, alveolar epithelial cells are plausible producers of citrullinated peptide [32]. Interestingly, Padi2 and Padi4 were detected in both respiratory epithelia and invasive epithelial cells (figure 5b). Only Padi4 was expressed in the respiratory epithelium at week 0, but not in DBA1/J mice, suggesting that Padi4 expression was upregulated in D1CC×D1BC mice (supplementary Figure 6). Recently, a similar characteristic basaloid cell, which expresses COL1A1 and MMP7 but no Krt5, was identified by single cell analysis [33, 34]. Since aberrant basaloid cells uniquely expressed KRT17, we investigated whether Padi4-positive cells express Krt17. We identified a Padi4-, Krt17-positive invasive population in vehicle, but there is no double-positive population in nintedanib-treated mice and age-matched controls (figure 5c). Nintedanib reduced the expression of Padi4 in the respiratory epithelium. Moreover, nintedanib also decreased citrullinated peptides (figure 5d). To further confirm the expression of Pad4 in the whole lung, we performed Western blotting. The expression of Pad4 was reduced by the administration of nintedanib (figure 5e).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Nintedanib suppressed the expression of Padi4 and the production of citrullinated peptide in respiratory epithelia. a) In situ hybridisation of Scgb1a1 (green), Tgf-β (white) and Col1a1 (red). Tgf-β and Col1a1 express in both alveolar epithelium and invasive epithelial cells. As nintedanib suppressed the occurrence of invasive epithelial cells, Tgf-β and Col1a1-positive invasive cells are not observed. b and c) In situ hybridisation of Scgb1a1 (green), Padi4 (white) and Padi2 or Krt17 (red). Padi4 and Padi2 expression is reduced in nintedanib treatment. d) Immunohistochemical analysis using E-cadherin (Ecad, green) and peptidyl-citrulline (red) in vehicle- or nintedanib-treated and age-matched mouse lungs. Scale bars=50 μm (white) and 20 μm (yellow). e) The expression of Pad4 in the lung tissues is assessed by Western blotting. Data are presented as mean±SE of four mice for each group. *p<0.05 compared with vehicle group.
Discussion
A novel iRA-ILD model using D1CC×D1BC mice developed NSIP with partial UIP as a histopathological feature. Anti-fibrotic nintedanib suppressed fibrosis and eliminated invasive epithelial cells. Finally, nintedanib reduced the expression of Pad4 and citrullinated peptides in the lung, resulting in the suppression of EMT in epithelial cells.
The inflammatory arthritis model is described in conventional animal models such as collagen-induced arthritis; however, it does not reflect chronic symptoms seen in RA and pulmonary fibrosis as a complication. In this study, we demonstrated that chronic progressive joint disorder and subsequent pulmonary fibrosis were observed in >60% of D1CC×D1BC mice. There was no upregulation of serum SP-D levels during inflammatory arthritis. The pathogenic mechanisms of RA-ILD are poorly understood, but high levels of serum RF IgG and IgM and anti-citrullinated protein antibodies may function as plausible risk factors for the development of ILD [30]. D1CC×D1BC mice may be highly susceptible to developing RA-ILD because of de novo citrullinated peptide by PADs in the respiratory epithelium. Nintedanib reduced the expression of Padi2, Padi4 and citrullinated peptides in the respiratory epithelium. This may reduce the production of target peptides by ACPA. The direct effects of nintedanib on antibody-producing plasma B-cells have not yet been described, so details of biochemical and pathological studies on the production of ACPA are needed. This helps clarify the intervention via ACPA between RA and subsequent ILD.
Nintedanib reduced the number of infiltrated M2 macrophages and inhibited M2 polarisation. Adhesion via Type I collagen activates PI3 K, followed by phosphorylation of Akt, which causes M2 macrophage polarisation. This effect was attenuated by the PI3 K inhibitor [35]. Nintedanib has been shown to reduce phospho-Akt expression [36] and may result in a decrease in the number of M2 macrophages. Thus, the anti-inflammatory effects of nintedanib could suppress fibrosis by inhibiting the production of Th2 cytokines via M2 macrophages. Serum cytokines, including Th2 cytokines, were reduced marginally. To this end, we should measure cytokines in bronchoalveolar lavage (BAL), but not in serum. However, measurement of cytokines in BAL and histopathological analysis is technically incompatible, which is a limitation of our study.
Complex pathological features in ILD lead to various histopathological classifications, suggesting that a single patient has a mixed pattern of UIP and NSIP [37]. Invasive epithelial cells were observed in the iRA-ILD mouse model. At this time, no morphological tumorigenic epithelial cells were found, except for some acquired EMT-related features such as the expression of Col1a1 and Tgf-β. We also observed that some invasive epithelial cells were Padi4- and Krt17-positive. Further research will elucidate the potential for EMT-mediated tumorigenesis and its relation to aberrant basaloid cells in the iRA-ILD model. The therapeutic effects of nintedanib on these epithelial cells are controversial [38, 39], but nintedanib is a potent inhibitor of tumorigenesis by blocking the expression of EMT-related molecules Tgf-β, Padi2 and Padi4 in respiratory epithelia.
Various functional blockages by nintedanib against invasive epithelial cells, respiratory epithelia and M2 macrophages, as well as fibroblasts, may suppress lung fibrosis. This is because nintedanib has a wide range of inhibitory effects on the development of RA-ILD. In fact, structure-based analysis and kinetic inhibition assay by nintedanib result in the blockade of at least 34 kinases, including PDGFR, FGFR and VEGFR. There are also various inhibitory activities for epithelial cell-related kinases such as Abl1, Kit, TRAK and Ret. Genome-scale analysis of lung cancer also identified a couple of prognostic biomarkers including kinases [40]. Inhibitors of these various kinases have the potential to be therapeutic agents for RA-ILD.
The effects of nintedanib extend not only to fibroblasts but also to macrophages and epithelial cells, resulting in reducing the overall symptoms of ILD in the mouse model. This evidence may support improved treatment in patients with RA-ILD, as nintedanib acts as a broad inhibitory effect on many tyrosine kinases.
Acknowledgements
We thank a Makoto Murata for his cooperation in measuring serum surfactant protein D and Stefan-Lutz Wollin for useful discussions. We also thank Boehringer Ingelheim Pharma GmbH & Co. KG for supplying nintedanib. We would like to thank Editage (www.editage.com) for English language editing.
Footnotes
Support statement: This work was supported by grants-in aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/JSPS KAKENHI Grant Number JP 26461470, 23591444, 17K09982, and 17K16055, research grant from Nagoya City University Grant Number 1943005. Funding information for this article has been deposited with the Crossref Funder Registry.
Author contributions: S. Kanazawa and H. Ohkubo were involved in the design of the study. Y. Miura, H. Ohkubo, and S. Kanazawa were involved in data acquisition and the analysis. All authors were involved in the interpretation of the data, drafted the final version of the manuscript.
Conflict of interest: S. Kanazawa and H. Ohkubo have received research support from Boehringer Ingelheim Pharma GmbH & Co. KG. S. Kanazawa has received personal donation from T. Furuya.
Ethics approval: All mouse experiments were performed according to the rules and regulations of the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology Japan and approved by the committee on the ethics of Animal Experiments of Nagoya City University.
Provenance: Submitted article, peer reviewed.
This article has supplementary material available from openres.ersjournals.com
- Received July 5, 2021.
- Accepted August 19, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










