





Abstract
Patients with progressive fibrosing interstitial lung diseases (fILD) have increased morbidity and mortality. Lung fibrosis can be associated with lung cancer. The pathogenesis of both diseases shows similarities, although not all mechanisms are understood. The combination of the diseases is challenging, due to the amplified risk of mortality, and also because lung cancer treatment carries additional risks in patients with underlying lung fibrosis. Acute exacerbations in fILD patients are linked to increased mortality, and the risk of acute exacerbations is increased after lung cancer treatment with surgery, chemotherapy or radiotherapy. Careful selection of treatment modalities is crucial to improve survival while maintaining acceptable quality of life in patients with combined lung cancer and fILD. This overview of epidemiology, pathogenesis, treatment and a possible role for antifibrotic drugs in patients with lung cancer and fILD is the summary of a session presented during the virtual European Respiratory Society Congress in 2021. The review summarises current knowledge and identifies areas of uncertainty. Most current data relate to patients with combined idiopathic pulmonary fibrosis and lung cancer. There is a pressing need for additional prospective studies, required for the formulation of a consensus statement or guideline on the optimal care of patients with lung cancer and fILD.
Abstract
Lung fibrosis can be associated with lung cancer. More and better-designed studies are needed to determine the true incidence/prevalence of lung cancer in fILD. Optimal treatment strategies urgently need to be defined and evaluated. https://bit.ly/37CzTMu
Introduction
Interstitial lung diseases (ILDs) are characterised by pathological changes in the pulmonary parenchyma, sometimes triggered by inflammation, but sometimes with an epithelial–fibrotic pathogenesis, in which inflammation is believed to play little part. Idiopathic pulmonary fibrosis (IPF) is the most common type of fibrosing ILD (fILD); almost always progresses (at a highly variable rate); and has the worst prognosis. A histological pattern of usual interstitial pneumonia (UIP) can be diagnostic for IPF in the absence of other causes. A definite UIP pattern on computed tomography (CT) is reliably predictive of a UIP pattern at biopsy, and is characterised by honeycombing, reticulation, traction bronchiectasis and subpleural and basal predominance [1]. A UIP pattern in IPF and other disorders tends to progress [2]. Other fILDs can develop a progressive phenotype. Once progression despite management has occurred, the disease course is usually similar to IPF [3]. Acute exacerbations are acute flares of ILD, sometimes overtly triggered, e.g. by infection or surgery and associated with high short-term mortality [4]. Acute exacerbations can also increase mortality in patients with other fILDs [5]. Antifibrotic drugs have been approved for IPF and, recently, for other progressive fILDs that decelerate disease progression and thus increase survival [1, 6–8]. Increased survival due to delayed disease progression in IPF and other progressive fILD patients leads to an increased prevalence of comorbidities and complications, with their optimal management complicated by concurrent ILD. Lung cancer as a comorbidity in ILD patients is the focus of the present review.
Epidemiology and risk factors of lung cancer in ILD patients
Lung cancer is an important comorbidity encountered in ILD patients, especially in IPF [9]. In some statements, confusion has arisen due to failure to distinguish between prevalence, incidence and cumulative incidence. In the following summary, we present an overview of current knowledge, emphasising the importance of these distinctions. Lung cancer prevalence refers to the total number of people suffering from lung cancer at a single point in time in a study population [10]; lung cancer incidence refers to the rate of development of new cases during a short time period (usually expressed as annual incidence) [10]; lung cancer cumulative incidence is the total proportion of patients developing lung cancer during a prolonged period of time (e.g. 5 years) [11].
Prevalence
A review by Ballester et al. [12] reported that the prevalence of lung cancer in IPF ranges from 2.7% to 48% and is significantly higher than in the general population. A meta-analysis has shown difference by region in prevalence of lung cancer in fILD. The prevalence in Asian cohorts is 15.3% and in European cohorts is 11.6% [13].
Incidence
Overall cancer incidence in IPF was 290 cases per 10 000 person-years in a population-based cohort study from Korea with 25 241 IPF patients and 75 723 matched controls. Risk of lung cancer was the highest, followed by lymphoma and skin cancer [14]. In a study from the United Kingdom studying incidence of lung cancer in IPF, there was an increased incidence compared to the general population (rate ratio 4.99) due to a marked increase of lung cancer incidence in IPF patients (112 per 10 000 person-years in IPF compared to 22.9 per 10 000 person-years in the general population) [15]. A study from the United States found that the incidence of lung cancer in IPF was 3.34-fold higher than in the general population [16]. This study also compared cancer laterality, primary site, histology and stage. They found statistically significant differences in lung cancer in IPF compared to lung cancer in a general population. This suggests that lung cancer in IPF is phenotypically distinctive from “sporadic” lung cancer [16]. However, in patients with ILD and rheumatoid arthritis, polymyositis/dermatomyositis or systemic sclerosis, the increase was as high as 4.95-fold [16]. Lung cancer origin and consequence can be additionally confounded in the face of risk factors such as smoking [17], silica and asbestos [18]. The histopathological UIP fibrosis pattern predisposes to pulmonary cancerous lesions [19]. Genetic factors contributing to cancer susceptibility and also predisposing to IPF include SFTPA1 and SFTPA2 [20].
Cumulative incidence
The cumulative cancer incidence rises strikingly with longer follow-up in IPF patients. In one study, the incidence of lung cancer was 1.1% at 1 year, 8.7% at 3 years, 15.9% at 5 years and 31.1% at 10 years [21]. A nationwide population-based study in Korea found the prevalence of lung cancer in IPF cases to be 6.4%. The median time from diagnosis of IPF to lung cancer development was 16.3 months [23]. The cumulative incidence of lung cancer in IPF increases from 1.7% at 1 year to 4.7% at 3 years and 7% at 5 years [22]. The incidence of lung cancer is increased in patients with ILD and COPD, but even more in patients with IPF or COPD and ILD [23].
Risk factors for cancer in fibrotic ILD are older age at diagnosis [24], smoking [24–28], male gender [25–28], IPF per se (adjusted for age, gender and smoking) [14], rapid annual decline in forced vital capacity (FVC) and low diffusing capacity of the lung for carbon monoxide (DLCO) [26, 28], as well as emphysema [27, 28].
Disease characteristics and prognosis of lung cancer in ILD patients
Lung cancer in IPF patients (LC-IPF) occurs more frequently in the lower lobes (p<0.001), whereas non-IPF ILD patients do not differ in localisation of lung cancer to the general population [16]. A clinicopathological study in Japan divided subjects into histological subgroups: UIP (clinically IPF group), non-UIP (clinically non-IPF ILD) and a normal group (without ILD), and showed that patients with lung cancer and UIP on histology had a subpleural lung cancer predilection in the lower lobes (75.5%), matching the typical distribution of UIP [19]. The most frequent cancer type was squamous cell cancer (62.8%) [19]. In comparison, in the non-UIP group, lung cancer occurred most frequently in the upper lobes (68.1%), with adenocarcinoma the commonest cancer type (55.2%) [19].
The occurrence of cancer within the fibrotic regions was confirmed by another study, in which lung cancer localisation within fibrotic areas was seen in 50%, followed by marginal-fibrotic areas (29%) and extrafibrotic areas (13%) [29]. Radiologically, lung cancer is most often found on CT in areas within fibrosis (44.4%) followed by adjacent areas abutting fibrosis (29.6%) [21].
In a large multicentre Greek cohort (n=1016), LC-IPF included squamous cell carcinoma in 34.3%, adenocarcinoma in 27.5% and small cell lung carcinoma (SCLC) in 14.7% in histological samples [30]. Again, 57% of patients had cancer within the lower lobes [30]. Squamous cell carcinoma was confirmed as the most frequent histopathological type in other studies [31]. Cancer-type distributions with squamous cell cancer in 37.8% of patients and adenocarcinoma in 30.8% of patients were found in a meta-analysis of a total of 131 947 IPF patients, of whom 6384 had lung cancer, from eight different countries [32].
Survival differences between IPF without lung cancer and IPF with lung cancer were statistically significant (p<0.001) [16], with lower survival over time for those with IPF and cancer [16, 27, 33]. A single-centre retrospective study found an increased mortality in patients with connective tissue disease associated ILD with lung cancer as compared to those without lung cancer [28].
Both the histological cancer type and its stage influences survival in patients with IPF, as in the general lung cancer population. Depending on the histological type and stage, outcome in IPF patients can be additionally impacted. In a recently published study in patients with lung cancer, LC-IPF patients had a poorer prognosis than a control group of lung cancer patients (5-year survival rate 14.5% versus 30.1%, p<0.001) [34]. The IPF subgroup had a worse prognosis than the group without IPF among patients with adenocarcinoma (median survival 11 versus 26 months, p<0.001) or squamous cell carcinoma (median survival 19 versus 30 months, p=0.003) [34]. The median survival of patients with lung cancer and IPF was shorter than that of the group without IPF in stage I (34 versus 77 months, p<0.001) and III of nonsmall cell lung carcinoma (NSCLC) (13 months versus 18 months, p=0.013). Median survival was similar in the IPF group and the group without IPF with stage II (23 months versus 28 months, p=0.142) and stage IV NSCLC (6 months versus 7 months, p=0.220) [34]. Among patients with SCLC, the median survival of the IPF and non-IPF groups was similar in both limited (16 months versus 16 months, p=0.456) and extensive stages of SCLC (6 months versus 9 months, p=0.379) [34]. In addition to cancer type and stage, morbidity and mortality are influenced by treatment modalities in all patients, but especially in patients with IPF and fILD.
Pathogenesis of cancer in ILD
The increased prevalence and accumulative incidence of lung cancer in fILD suggest that ILD may itself promote lung cancer development [14]. Specifically, in IPF, some molecular and genetic features of its pathogenesis and progression are linked to mechanisms that favour development of malignancy [12]. Unfortunately, the evidence for pathogenesis of lung cancer in ILD patients is vague. The following hypotheses are drawn from studies in patients with fILD or lung cancer. These findings need further confirmation in ILD patients with lung cancer and should be considered carefully.
Histopathological studies in IPF, the archetype of fILD with a UIP pattern as the histological background, suggest that abnormal bronchiolar proliferation in fibrotic areas might be the pre-neoplastic lesion [35]. An immunohistochemistry study of 33 cases of lung cancer arising in patients with IPF demonstrated that neoplastic cells express bronchial markers including the transcription factor-1, napsin-A and surfactant protein A, expressed also in a minority of cases of adenocarcinoma [35].
Proteins involved in cell renewal of bronchial epithelium such as ΔNp63 were found to be overexpressed in bronchioloalveolar junctions in IPF lungs [36].
Abnormal activation of the Wnt/β-catenin pathway was documented in fibrotic areas in lung samples obtained from patients with IPF, and this pathway could be involved in squamous dysplasia and in promoting squamous carcinoma differentiation [37, 38]. Recently, single-cell sequencing and gene expression analyses have supported histopathological findings of proliferative bronchiolar structures or bronchiolisation in lung samples with a UIP pattern [38]. Increased airway epithelial cells populations at the expense of the typical alveolar epithelial cell markers were found [39]. Moreover, airway basal cell populations (CK5/6+ and ΔNp63+) have been described in the surroundings of the fibroblast foci in a study of bronchoalveolar lavage cell expression in IPF patients [40].
One of the cardinal mechanisms that may promote bronchial pre-cancerous lesions in IPF lungs is alveolar epithelial cell exhaustion [41]. Both intrinsic (e.g. genetic, ageing) and environmental factors (e.g. smoking, pollution) contribute to alveolar stem cell dysfunction in patients with IPF. These cells express senescence markers and are unable to rebuild the lung parenchyma properly after endogenous or exogenous insults. However, they acquire a senescence-associated secretory phenotype inducing aberrant activation of important regulators of cell transformation, growth and migration signals and epithelial–mesenchymal transition (EMT) [37, 38] such as Wnt/β-catenin and Sonic Hedgehog pathways [42]. Along with formation of a fibrotic microenvironment [38], these processes lead to bronchial overgrowth and cancer cell development and progression [37]. In a subset of IPF patients, expression of membrane PD-L1 protein in alveolar and/or bronchiolar cells was documented, confirming the pathogenetic role of EMT and reinforcing the links between IPF pathogenesis and carcinogenesis [43].
Atypical squamous cells are frequently found in honeycombing areas. Serpin B4 overexpression in these metaplastic cells was related to both transforming growth factor-β and Ki-67 overexpression and was higher in patients with foci of cancer/high-grade dysplasia, showing that this pathway could be another important cofactor for cancer development in IPF lungs [44].
In addition, some genes under post-transcriptional control of miR-200 are overexpressed in bronchiolar fibroproliferative lesions of IPF lungs that are microRNAs regulating EMT and tumour cell adhesion. Proteins promoting abnormal migration of bronchiolar cells, such as laminin-5-γ2 chain and Hsp27 were found to be overexpressed in cells covering fibroblastic foci or honeycomb cysts; this further suggests that activation of EMT might have a role in abnormal bronchiolar progression in these areas [45, 46].
Along with these typical fibrosis-related mechanisms, a genomic study carried out by Hwang et al. [47] has provided a new perspective on the origin of lung cancer in ILD. The genomic profile of lung cancer associated with IPF showed a significantly higher prevalence of mutations in TP53 and BRAF in their cohort, genes implicated in cell proliferation and survival, suggesting a genetic susceptibility to lung cancer in patients with IPF [47].
In summary, bronchiolar hyperplastic–dysplastic cells are possibly the driver of lung cancer in ILD. Bronchiolisation of distal areas within UIP patterns is promoted by a profibrotic microenvironment, and the mechanisms involved in fibrosis development may also activate molecular processes able to induce pre-neoplastic, and in more advanced stages, cancerous lesions, as shown in figure 1.
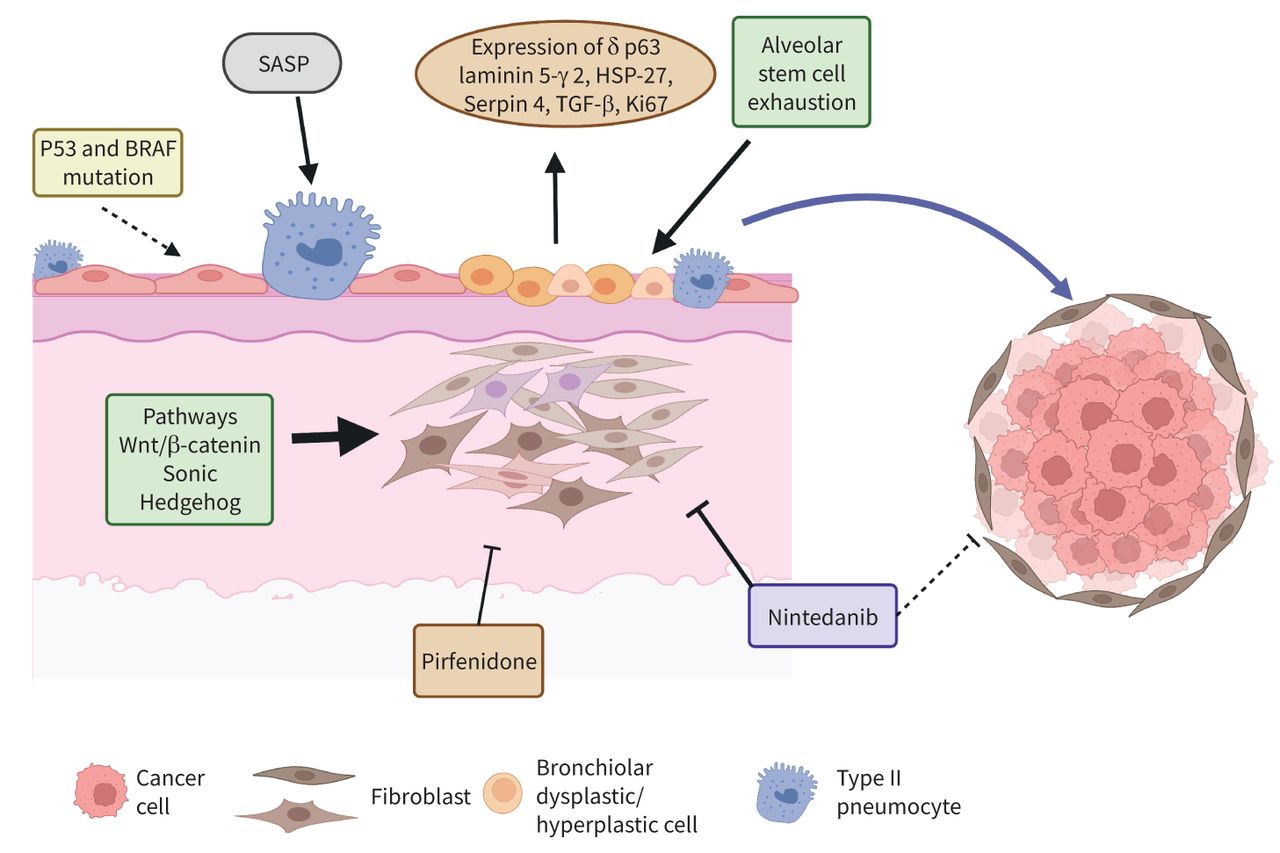
Pathomechanisms of lung cancer and fibrosing interstitial lung disease. Dysplastic bronchial cells accumulate near fibroblastic foci. Senescence-associated secretory phenotype (SASP) and stem-cell exhaustion contribute to dysplastic bronchial cell development. Fibrotic processes including epithelial–mesenchymal transition, Wnt/β-catenin and Sonic Hedgehog pathways contribute to cancer development. Antifibrotic drugs inhibit fibrosis and might have potential effects on cancer. HSP: heat shock protein; TGF: transforming growth factor. Created with BioRender (www.biorender.com).
Treatment of lung cancer in ILD patients
Lung cancer treatment is currently tailored to each individual according to the stage, type of cancer and the performance status of the patients. Lung cancer in ILD has some peculiarities which pose a challenge to care providers, especially in IPF patients. Patients with IPF are typically of older age, often smokers and highly comorbid. The underlying ILD leads to reduced lung functional performance and respiratory capacity (i.e. reduced FVC and DLCO). Mortality is increased with increasingly severe lung function impairment, as judged by FVC, DLCO, the Composite Physiologic Index or the ILD–Gender Age Physiology (GAP) index. In addition, underlying ILD predisposes to a risk of acute exacerbation and, thus, increased mortality [33]. Therapeutic strategies for lung cancer in patients with fILD need to be adapted according to the individual treatment risk and the prognosis of both lung cancer and underlying ILD [48].
Early and late lung cancer mortality in patients with ILD is increased after adjustment for confounding factors. Some of the reasons for early mortality are acute exacerbation of the fILD related to surgery, irradiation or anticancer drugs [49]. Late mortality may be due to lung cancer progression or relapse, and also to ILD natural history [50]. Treatment options for NSCLC have improved dramatically over the past decade. An individual and personalised treatment approach to lung cancer in patients with fILD is desirable.
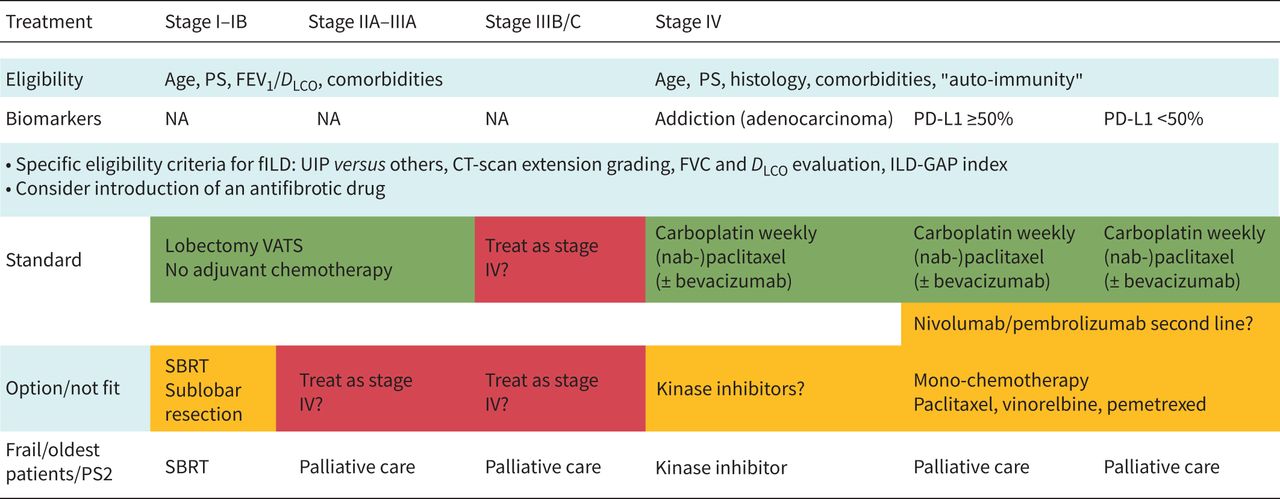
An overview of modern therapeutic strategies for lung cancer with fILD other than SCLC is illustrated in figure 2 and suggestions are summarised in table 1. Next to surgical treatments for selected candidates, stereotactic body radiation therapy (SBRT) can be chosen for frail patients with early-stage cancer. Combination treatments or only chemotherapy/targeted anticancer drugs might be chosen for more advanced stages, depending on overall individual frailty and age. The individual treatment options for SCLC and NSCLC are described later.

{kind=link}
{kind=link}
A proposal for adapted modern therapeutic strategies for lung cancer fibrosing interstitial lung disease (fILD) other than small cell lung carcinoma. PS: performance status; FEV1: forced expiratory volume in 1 s; DLCO: diffusing capacity of the lung for carbon monoxide; NA: not available; UIP: usual interstitial pneumonia; CT: computed tomography; FVC: forced vital capacity; GAP: gender, age, physiology; VATS: video-assisted thoracoscopic surgery; SBRT: stereotactic body radiotherapy.
Suggestions for treatment of lung cancer in fibrosing interstitial lung disease (fILD)
Small cell lung cancer in fILD
Patients with SCLC and fILD respond well to standard treatment if they can tolerate chemotherapy. The prognosis of SCLC in patients with ILD is comparable to those without ILD [48].
In SCLC patients receiving chemotherapy, overall survival in patients with ILD was not inferior to that in patients without ILD. In ILD patients with an UIP pattern, the overall survival was reduced compared to non-UIP, although this was not statistically significant [51].
Early-stage NSCLC in fILD (stage I and II)
Surgery
Surgical options in NSCLC include lobar resection or anatomical sublobar resection, pneumonectomy or wedge resection depending on the tumour localisation, size, stage and morbidity. Lobar resection has a better prognosis than partial resection in early disease. In carefully selected patients, outcome can be improved by adjuvant chemotherapy. The factors determining better outcomes are lower stages than pIIA–IIIA (according to the eighth tumour, node, metastasis classification) [52], better performance status score, younger age and absence of comorbidities [53, 54]. In these situations, mortality at 5 years is reduced by 5–15%. In real life, <60% of general lung cancer patients receive adjuvant treatment [55, 56]. When surgery is not possible due to poor performance status score, early-stage NSCLC (tumour <3 cm) can alternatively be treated by SBRT [57].
Peri-operative mortality is increased in IPF patients undergoing cancer surgery [58]. Post-operative and surgery-related mortality are increased in IPF with a lower 5-year survival after pulmonary resection of NSCLC in IPF compared to non-IPF [59]. The 5-year survival in patients with NSCLC was 43% in patients with IPF and 64.2% among those without IPF (p<0.001). Disease-free survival was similar in the groups [60]. In a matched case–control study from Korea, 33 patients with IPF who had undergone surgery were matched with 66 control patients who had undergone lung cancer surgery. The 5-year survival rate was 38% for lung cancer patients with IPF and 73% for the control group (p=0.001) [61]. In a Japanese study with 870 lung cancer patients undergoing surgery, 56 patients had IPF. Surgery-related mortality was higher in patients with lung cancer and IPF than in patients with lung cancer alone (7.1% versus 1.9%, p=0.030) [58]. One reason for increased mortality associated with surgery is acute ILD exacerbations, addressed later in the discussion of the role of antifibrotics in lung cancer treatment.
Radiotherapy
For patients with poor lung reserve or comorbidities, even in early NSCLC, surgical and chemotherapeutic options are limited. In a systematic review by Chen et al. [57] of patients with early-stage NSCLC and ILD, high levels of treatment-related toxicity and ILD-specific toxicity were documented in patients undergoing SBRT, particle beam therapy or radiofrequency ablation. Survival without treatment was 12 months. The pros and cons of undergoing potential toxic treatment versus best supportive care must be carefully considered by both clinicians and patients. A retrospective study by Onishi et al. [62] in 242 patients with ILD and early-stage lung cancer receiving SBRT found the rate of severe radiation pneumonitis to be 12.4%. The mortality rate was 6.9%. Some of the risk factors for poor outcome include FVC <70%, >10% of normal lung receiving radiation, performance status 2–4, presence of squamous cell carcinoma and clinical stage T2, as well as regular use of steroid before SBRT [62]. Very few data evaluate the efficacy and toxicity of conventional radiation therapy in ILD patients with lung cancer (LC-ILD). Radiotherapy-induced pneumonitis can add to mortality in LC-IPF and contributes to the overall poor outcome in these patients [63]. The European Organisation for Research and Treatment of Cancer recommends that conventional radiotherapy should be avoided for patients with LC-IPF [64].
Late-stage NSCLC
In advanced stages, lung cancer can be treated with chemotherapy, targeted therapy and immunotherapy, in general. The few prospective or case–control cohorts contain limited numbers of patients (15–100) with advanced LC-fILD, among whom IPF is present in 25–100% of cases. Almost all studies were conducted with carboplatin [65–70]. Most studies evaluated carboplatin in combination with weekly paclitaxel (or nanoparticle albumin-bound, nab-paclitaxel) [71–75], with fewer data on immune checkpoint inhibitors (ICIs) [70, 76]. The results from these studies need to be interpreted with caution. The limited database shows an unexpectedly high proportion of response rate (27–70%) in the previously cited studies. Response criteria such as tumour shrinkage have been defined to standardise response rate assessment [77]. In the studies, patients had good performance status; the histology was variable; and a high proportion of stage IIIA–B patients received chemotherapy instead of surgery/radiotherapy, enriching the population of “good prognosis” cancer patients. In addition, they did not include patients with poor lung volumes or low DLCO. The progression-free survival was 3.7–7.2 months, with an overall survival of 5.4–19.4 months. Acute exacerbation was observed in 2.8–12% of cases.
Chemotherapy, kinase inhibitors and ICIs
In retrospective studies, the overall survival was increased in lung cancer patients (with or without ILD) receiving chemotherapy compared to patients who received palliative care, as shown in a cohort studying the effects of chemotherapy in patients with LC-ILD (specifically idiopathic interstitial pneumonias) [51]. Median survival time (MST) was 25.0 versus 1.8 months (p<0.001). In patients receiving chemotherapy, the overall survival was reduced in ILD patients (MST 10.9 months versus 25.0 months, p=0.043). In NSCLC, the overall survival was reduced in patients with ILD (MST 10.6 versus 27.9 months, p=0.008) [51].
No randomised prospective controlled trial has evaluated the effects of chemotherapy on lung cancer outcome in patients with fILD. In real life, a lot of LC-fILD patients are of advanced age and have various comorbidities, increasing the risks of treatment. Acute exacerbation of ILD is an important complication of treatment. Thus, treatment options need to be tailored for individual cases, balancing individual risks and benefits. Acute exacerbation induced by chemotherapy in advanced LC-IPF increases the risk of mortality [69]. In a Japanese study of 69 patients, those with a UIP pattern on chest CT scan developed acute exacerbation after chemotherapy more frequently than those with a non-UIP pattern (30% versus 8%, p=0.005) [69]. A study by Kanaji et al. [78] observed that patients with ILD and IPF treated with paclitaxel/nab-paclitaxel had few acute exacerbations. Acute exacerbations in patients treated with docetaxel were seen in 18.4% and 20.8% of patients with ILD and IPF, respectively.
Two retrospective cohorts suggested that adding bevacizumab reduces the risk of ILD progressions or acute exacerbation, even if ILD was related to chemotherapy [70, 79]. Bevacizumab is a monoclonal antibody against vascular endothelial growth factor (VEGF) and has been used for NSCLC in combination with paclitaxel [80]. Interestingly, VEGF plays a role in ILD progression, and fibrosis development could be inhibited in a pre-clinical model of VEGF-A deficient alveolar type II cells in mice [81]. Of note, nintedanib, used to reduce IPF progression, is a well-known inhibitor of the receptor tyrosine kinase VEGFR [82].
In addition to standard chemotherapy, kinase inhibitors are now standard of care for advanced NSCLC with oncogene addiction [83]. However, drug-induced ILD is more frequent with kinase inhibitors than with chemotherapy. Prior ILD is a risk factor for kinase inhibitor-associated ILD. Mortality rate for kinase inhibitor-induced ILD is high [84]. In the general population, ICI are the standard of care in second line for advanced NSCLC, with pembrolizumab, atezolizumab and cemiplimab being standard of care as first-line treatment for advanced NSCLC with PD-L1 expression >50% or in combination with a double platinum chemotherapy in other fit patients (pembrolizumab and atezolizumab). In a phase II trial on nivolumab for advanced NSCLC in fILD, the 6-month progression-free survival rate was 56%, response rate was 39% and the disease control rate was 72% [85] with a low frequency of acute exacerbation and no treatment-related deaths. By contrast, another phase II trial with atezolizumab showed an incidence of pneumonitis in patients with fILD of 29.4%, leading to early study closure. However, in this trial, the proportion of IPF patients was high. The objective response rate was 6.3% [86]. A retrospective study confirmed increased incidence of pneumonitis in patients receiving nivolumab [87]. A retrospective study from Japan comparing nivolumab versus pembrolizumab showed noninferior outcomes with respect to progression-free survival and overall survival [88]. In summary, ICI studies are scarce, mostly consisting of retrospective cohorts, with contradictory results, limited numbers of patients, confined to Asian countries and mostly investigating patients with IPF.
Palliative care
In addition to tumour-directed treatments, palliative care should be considered early in the treatment process. Palliative care medicine is often wrongly perceived as a terminal-phase treatment [89]. It is centred on patient needs, providing comfort and symptom control.
fILD and lung cancer are each fatal in isolation and their combination has a synergistic effect in increasing mortality and reducing quality of life. A review published by Naccache et al. [48] summarises this grim scenario. In patients with lung cancer and fILD after resection, a second lung cancer is observed in 36% of cases. In combined pulmonary fibrosis and emphysema (CPFE), 43% of patients with lung cancer do not receive standard care because of underlying CPFE. In advanced fILD, 20–25% of patients are unable to receive chemotherapy due to frailty, and 50% of patients receive only one type of chemotherapy [48].
Role of antifibrotics in ILD and cancer
Antifibrotics reduce acute exacerbation due to cancer treatment
Cancer treatment-associated mortality, especially in patients undergoing surgical resection, can be attributed to acute exacerbation. The incidence of post-operative acute exacerbation was 6.4% in a single centre study from Tokyo [90]. In a multicentric data analysis from Japan, post-operative acute exacerbation of ILD occurred in 164 (9.3%) patients with an overall mortality rate of 44% [91]. The timing peak of acute exacerbation was at day 4. 64% developed acute exacerbation in the first 10 days post-surgery [91].
Risk factors for acute exacerbation after surgical resection include type of surgery, elevated KL-6 levels, male sex, reduced vital capacity (%), history of acute exacerbation, pre-operative steroid use and a UIP pattern on CT [48, 92]. In patients with LC-ILD, surgical procedures have shown the strongest association with acute exacerbation, possibly due to handling of the lungs, lymphatics and vasculature during the intervention [91]. The risk of acute exacerbation increases according to the volume of lung removed, with the highest risk for pneumonectomy and lowest risk for wedge resection. Minimally invasive surgery such as video-assisted thoracoscopic surgery does not reduce the risk of acute exacerbation [93]. Controlling intra-operative intravenous fluid has been shown to be protective for development of acute exacerbation in IPF patients undergoing lung cancer surgery [94]. A single-centre study from Japan found that high oxygen concentration with single-lung ventilation and hyperventilation with high airway pressure increased the risk of acute exacerbation in idiopathic interstitial pneumonia patients undergoing surgical resection [95].
In a retrospective review of pre-operative CT and histopathological examination in patients who underwent resection for lung cancer, the incidence of clinical acute respiratory distress syndrome (ARDS; synonymous with acute exacerbation in ILD patients), was 31.8% in patients with “interstitial pneumonia” defined histologically, which was strikingly higher than the 1.5% prevalence observed in the “interstitial pneumonia-negative group” [59]. This suggests that the histopathological presence of interstitial pneumonia trumps other risk factors for post-operative ARDS, manifesting clinically as breathlessness and histologically as diffuse alveolar damage [96]. An observational study by Oishi et al. [97] suggests that high maximum standardised uptake values in positron emission tomography imaging in idiopathic interstitial pneumonia areas may predict both acute exacerbation after lung resection and short-term survival. Ueno et al. [98] noted that ILD GAP index can predict prognosis in patients with lung cancer and ILD undergoing surgical resection.
The benefits of antifibrotic agents in patients with LC-ILD
Antifibrotic drugs have been observed to reduce acute exacerbation in IPF. In the INPULSIS trials of nintedanib in IPF, the frequency of acute exacerbation was reduced in the active treatment arm [99], although data on this potential benefit of pirfenidone are less conclusive in the general IPF population. Paradoxically, studies of the benefits of antifibrotic agents in reducing acute exacerbation prevalence in LC-ILD patients following cancer interventions are mostly confined to pirfenidone.
The potential efficacy of peri-operative pirfenidone in reducing the incidence of post-operative acute exacerbation IPF and thus reducing post-operative mortality, has been explored in retrospective cohorts of lung cancer patients undergoing resection surgery (summarised in table 2) [100–103]. These studies have several limitations. Underpowered retrospective cohorts are subject to publication bias and to potential differences in standard of peri-operative and post-operative care between active and inactive arms if open therapy is used. In two studies, a rigorous protocol for the duration of pre-operative pirfenidone was used, raising the possibility that the whole operative protocol was more rigorous in this patient subgroup. It is clearly possible that baseline difference between treated and untreated groups might have separately influenced post-operative outcomes. Therefore, while the data from these studies are suggestive of a pirfenidone protective effect, a prospective placebo-controlled study is required if peri-operative pirfenidone is to become standard of care. It should be noted that all three studies were conducted in Japan, with the possibility that genetic factors might limit the generalisability of the findings. Nintedanib has not been studied in this context, as pirfenidone has been the antifibrotic therapy routinely used in Japan. In addition, nintedanib has potential bleeding side-effects due to anti-angiogenic properties that might complicate cancer treatment [104].
Core features of studies of post-operative outcomes, comparing patients treated and not treated with peri-operative pirfenidone (poPirf)
There is some evidence that antifibrotic therapy reduces the risk of radiation pneumonitis in animal studies, but there are no compelling human data. In animal experiments, a study by Sun et al. [105] found that oral pirfenidone prevents radiation-induced interstitial fibrosis when administered in rats. A study in 266 mice showed that nintedanib administration diminishes histological signs of radiation-induced lung damage [106]. A study by Qin et al. [107] observed that pirfenidone also protects against radiation-induced pulmonary fibrosis in mice. Human data are needed to establish proof of concept before a definitive trial.
Data are beginning to emerge that antifibrotic therapy may facilitate chemotherapy by reducing complications of chemotherapy. The prevalence of acute exacerbations with chemotherapy in lung cancer/IPF is estimated to be 10–30%. A study with carboplatin plus nab-paclitaxel with and without nintedanib was designed to demonstrate that addition of nintedanib prolongs the interval to acute exacerbation [108]. The results of this trial are expected soon. In a study of 14 IPF patients with NSCLC receiving first-line chemotherapy (carboplatin+paclitaxel) in combination with pirfenidone, no acute exacerbations or adverse safety signals were reported [109]. Although there are currently insufficient data to establish proof of concept, the potential importance of these observations fully justifies a high priority for larger prospective studies.
Direct effects of antifibrotics on cancer
Over the past decade, the treatment of fibrotic ILD has been revolutionised by the use of antifibrotic drugs that reduce fibrosis progression in IPF and other progressive fibrosing ILDs [110]. Pirfenidone and nintedanib are currently the only approved antifibrotic drugs, with additional candidate antifibrotic therapies undergoing clinical trial evaluation. As the pathogenesis of ILD and lung cancer includes overlapping pathways, it is theoretically possible that antifibrotic therapy has antineoplastic effects [12]. Interestingly, nintedanib in association with docetaxel is an approved second-line cancer drug for selected NSCLC patients and has been used in patients with IPF and cancer [111].
Several reports show that pirfenidone targets pathways implicated in lung cancer pathogenesis. Pirfenidone inhibits cancer fibroblasts, and enhances ICI efficacy in mice [112]. In vivo studies confirm targeting of cancer fibroblasts and inhibition of fibroblasts and stroma cross-talk [113]. Pirfenidone induces cell cycle arrest in human and mouse cells and inhibits cancer proliferation [114]. In vivo, it interferes with the urokinase system and may influence the stability of tumour blood cells [115]. In vivo, it may revert EMT in lung adenocarcinoma [116]. However, human data are required for proof of concept (table 3).
Current role of antifibrotic drugs in lung cancer and fibrosing interstitial lung diseases (fILD)
Retrospective study data from 261 IPF patients with and without pirfenidone showed lung cancer incidence of 2.2% in the pirfenidone group and 22% in the non-pirfenidone group (p<0.001) [117]. On multivariable analysis, lung cancer incidence was lower in patients treated with pirfenidone (hazard ratio (HR) 0.11, p=0.003) and higher in patients with concurrent emphysema (HR=3.22, p=0.009) [117]. However, these data, if confirmed, do not indicate that pirfenidone prevents cancer genesis. Lung cancer manifests as a pulmonary nodule after perhaps 30 tumour doubling times. If we assign average doubling time as 4–6 months, it follows that lung cancer is present for >10 years by the time it is diagnosable [118]. Therefore, the data suggest that pirfenidone may slow cancer progression before it is clinically detectable. If so, it is possible that the lengthier survival achieved by antifibrotic therapy will not necessarily result in a major increase in the lung cancer burden.
The use of antifibrotic therapy for ILD in fILD patients with lung cancer
In untreated patients diagnosed with lung cancer and ILD, there is no evidence that use of antifibrotic agents should differ from their use in ILD in general. If lung cancer is advanced, without the option of radical interventions, and the approach is broadly palliative, the introduction of antifibrotic drugs with the goal of slowing ILD progression is unlikely to be helpful and may reduce quality of life. However, this scenario aside, there are no data to suggest that ILD management should be modified. When a definite or working diagnosis of IPF is made, antifibrotic therapy may improve life expectancy with the added possibility that acute exacerbation triggered by resection surgery, chemotherapy or radiotherapy may be reduced in prevalence, as discussed earlier. When an alternative diagnosis to IPF is made, and ILD is overtly progressive, the documented benefits of antifibrotic therapy in progressively fibrotic ILD justify its use [7, 110, 119]. It should be acknowledged that patients with fibrotic ILD associated with lung cancer were not included in these trials. However, the uniformity of treatment effects across a wide variety of non-IPF disorders in the INBUILD nintedanib trial can reasonably be extrapolated to this group of patients. There are no data to suggest that antifibrotic therapy in patients with pre-existing ILD should be discontinued when lung cancer is diagnosed. However, non-IPF patients managed with immunosuppressive therapy should ideally be discussed, case by case, with an oncologist, in view of the possible deleterious effects of these treatments in promulgating cancer progression.
Conclusion and outlook
More and better-designed studies are needed to determine the true incidence/prevalence of lung cancer in fILD. Optimal treatment strategies need to be defined and evaluated. The development of centres of excellence for ILD and cancer has the potential to improve patient care. As most studies included IPF patients, future studies need to include connective tissue disease associated ILD and other ILDs.
Footnotes
This article has been revised according to the correction published in ERJ Open Res 2022; 8: 50115-2022 [https://doi.org/10.1183/23120541.50115-2022].
Provenance: Commissioned article, peer reviewed.
Conflict of interest: N. Kewalramani reports grants and nonfinancial support from CSL Behring outside the submitted work.
Conflict of interest: C. Machahua has nothing to disclose.
Conflict of interest: V. Poletti reports personal fees from Boehringer Ingelheim, Roche, AMBU and ERBE, outside the submitted work.
Conflict of interest: J. Cadranel reports fees for participation on boards of experts for the development of cancer drugs from AbbVie, AZ, BI, BMS, Jansen, MSD, Novartis, Pfizer, Roche and Takeda.
Conflict of interest: A.U. Wells reports personal fees and nonfinancial support from Boehringer Ingelheim, Bayer and Roche Pharmaceuticals, and personal fees from Blade, outside the submitted work.
Conflict of interest: M. Funke-Chambour reports grants from Boehringer Ingelheim and Roche, and other support from MSD, outside the submitted work.
- Received March 1, 2022.
- Accepted April 18, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Epidemiology and risk factors of lung cancer in ILD patients
- Disease characteristics and prognosis of lung cancer in ILD patients
- Pathogenesis of cancer in ILD
- Treatment of lung cancer in ILD patients
- Role of antifibrotics in ILD and cancer
- Conclusion and outlook
- Footnotes
- References
- Figures & Data
- Info & Metrics