





Abstract
Bronchiolitis obliterans syndrome (BOS) may develop after either lung or haematopoietic stem cell transplantation (HSCT), with similarities in histopathological features and clinical manifestations. However, there are differences in the contributory factors and clinical trajectories between the two conditions. BOS after HSCT occurs due to systemic graft-versus-host disease (GVHD), whereas BOS after lung transplantation is limited to the lung allograft. BOS diagnosis after HSCT is more challenging, as the lung function decline may occur due to extrapulmonary GVHD, causing sclerosis or inflammation in the fascia or muscles of the respiratory girdle. Treatment is generally empirical with no established effective therapies. This review provides rare insights and commonalities of both conditions, which are not well elaborated elsewhere in contemporary literature, and highlights the importance of cross disciplinary learning from experts in other transplant modalities. Treatment algorithms for each condition are presented, based on the published literature and consensus clinical opinion. Immunosuppression should be optimised, and other conditions or contributory factors treated where possible. When initial treatment fails, the ultimate therapeutic option is lung transplantation (or re-transplantation in the case of BOS after lung transplantation) in carefully selected candidates. Novel therapies under investigation include aerosolised liposomal cyclosporine, Janus kinase inhibitors, antifibrotic therapies and (in patients with BOS after lung transplantation) B-cell-directed therapies. Effective novel treatments that have a tangible impact on survival and thereby avoid the need for lung transplantation or re-transplantation are urgently required.
Abstract
A review focusing on novel treatment approaches for bronchiolitis obliterans syndrome, to reverse the pathological changes and thereby impact tangibly on survival or need for subsequent lung transplantation, and improve patients’ quality of life https://bit.ly/3lCIB0x
Introduction
Pulmonary complications such as bronchiolitis obliterans syndrome (BOS) are common after lung or haematopoietic stem cell transplantation (HSCT) [1]. BOS is characterised by a progressive obstructive ventilatory defect due to the development of obliterative bronchiolitis [1–7]. Hitherto, the rarity of obliterative bronchiolitis delayed the understanding of its disease pathogenesis [8]. However, the emergence of lung transplant and HSCT as therapeutic modalities has led to an increased interest in this rare condition, providing much of the literature regarding the risk factors, natural history, therapeutic responsiveness and outcomes [9, 10]. While microvascular ischaemia likely plays a role in pathogenesis, the driving force is almost certainly immune related; for this reason, the twin pathways of “graft versus host” and “host versus graft” need to be compared.
BOS after lung transplantation is the most common phenotype of chronic lung allograft dysfunction (CLAD), developing in up to 50% of recipients by the fifth post-transplant year and is the major cause of late post-transplant mortality [11–13]. BOS after HSCT is less common, affecting 2–10% of recipients within 5 years [14–20]. BOS is the lung manifestation of multisystemic graft-versus-host disease (GVHD), with a prevalence of 14% in patients with chronic GVHD [21]. BOS negatively impacts prognosis after lung transplantation and HSCT [11–13, 18, 21–23]. However, as data required to confirm evidence-based therapeutic recommendations are limited, treatment is generally consensus driven [24].
The aims of the current article are to describe the characteristics of BOS after lung transplantation and HSCT, provide a viewpoint on diagnosis and management of these conditions based on expert opinion and a review of the literature, and describe research into new treatment strategies.
Methods
A videoconference of international experts (adult and paediatric transplant physicians, pulmonologists and haematologists), facilitated by a professional moderator, was held on 18 December 2020. This review summarises the consensus opinions of the group on BOS after lung transplantation and HSCT, supported by published literature.
What are CLAD-BOS and chronic GVHD-BOS?
CLAD
The International Society for Heart and Lung Transplantation (ISHLT) consensus statement defines CLAD as a persistent decline in pulmonary function, characterised by a decrease of ≥20% in forced expiratory volume in 1 s (FEV1) for >3 months from post-transplant baseline, after excluding other potential causes (table 1) [3]. The baseline value is calculated from the mean of the two best post-operative FEV1 measurements taken ≥3 weeks apart. CLAD severity is staged based on the current FEV1 relative to baseline [3].
Restrictive, obstructive, mixed or undefined clinical phenotypes of CLAD are defined based on the predominant ventilatory pattern, total lung capacity (TLC) and presence/absence of opacities on chest computed tomography (CT) scan (supplementary table S1) [3, 25]. Pulmonary function changes characteristic of the restrictive allograft syndrome (RAS) phenotype are declining TLC and a FEV1/forced vital capacity (FVC) ratio >0.7, whereas BOS is characterised by stable or increasing TLC and declining FEV1/FVC indicative of obstruction and hyperinflation [26]. The presence of multi-lobar persistent parenchymal and/or pleural opacities on CT (or chest radiograph (CXR) if CT is not available) is required for RAS diagnosis [3, 25].
∼65–70% of CLAD patients present predominantly with the BOS phenotype, and 10–35% have the RAS phenotype [12, 26–29], although the proportion with RAS appeared lower in a more recent study based on the latest ISHLT consensus criteria [29]. This is partly because mixed phenotype is now considered a different entity, whereas RAS and mixed were previously grouped together. Using these criteria, Levy and colleagues estimated that ∼5% of patients present with mixed and ∼10% with undefined phenotype [29], whereas Verleden and colleagues estimated that the undefined phenotype was less frequent (2%); however, raw data were not independently verified. Overall survival and graft survival are worse in those with RAS or mixed phenotype than the BOS or undefined phenotype [12, 26, 27, 29–31]. Survival differences between CLAD phenotypes are unaffected by age, sex, native lung disease and cytomegalovirus (CMV) serostatus mismatch [29]. However, allograft survival among all patients with CLAD is generally worse in those with CMV serostatus mismatch and in those failing to achieve predicted FEV1 and FVC post-operatively [12].
Phenotypes are not static, and patients can progress from the BOS to RAS phenotype, and more rarely from RAS to BOS [26, 32]. A patient who transitions from BOS to RAS will meet the criteria for the mixed phenotype [3, 25], so it is more accurate to describe the phenotypic change as BOS-to-mixed. Survival in BOS-to-RAS patients is worse than in BOS, but comparable to or better than in RAS patients [31, 33].
Risk factors for BOS and RAS include non-adherence to or suboptimal immunosuppressive regimens, acute cellular rejection (ACR) episodes, lymphocytic bronchiolitis, community-acquired respiratory viral (CARV) infection, donor-specific antibodies, air pollution, gastro-oesophageal reflux (GERD), Pseudomonas aeruginosa or Aspergillus fumigatus colonisation and CMV mismatch [31, 34, 35], although more RAS patients seem to develop human leukocyte antigen (HLA) antibodies compared with BOS patients [36]. Additionally, the development of RAS, but not BOS, appears to be delayed after living donor lung lobar versus cadaveric donor transplantation [37].
GVHD-BOS
GVHD-BOS usually develops between 100 days and 2 years of HSCT, but onset beyond 5–6 years post-HSCT has been noted, usually in patients experiencing an extrapulmonary GVHD flare [7, 16, 17]. Risk factors for GVHD-BOS are impaired lung function before and early after HSCT, a myeloablative/busulfan-containing conditioning regimen, CMV seropositivity, pre-transplant history of pulmonary disease, female donor, unrelated donor and prior acute GVHD; receipt of antithymocyte globulin, which decreases chronic GVHD risk, reduces the risk of BOS [17, 18, 38].
There is a need for clinical biomarkers at 80–100 days post-HSCT, because declining lung function at this time-point is a significant risk factor for later BOS development. Beyond chest irradiation and FEV1, forced expiratory flow between 25% and 75% of maximum has emerged as an important biomarker for the early detection of at-risk patients [38, 39].
Patients may present with persistent cough or dyspnoea-on-exertion, while asymptomatic patients may be identified during routine monitoring of pulmonary function [7]. The nonspecific symptomatology of GVHD-BOS may contribute to diagnostic delays [40]. Unlike CLAD, which affects only the lungs, GVHD is a systemic condition, so BOS after HSCT usually occurs in association with signs/symptoms affecting other organ systems, such as the skin, nails, eyes, mouth, hair, genitals, joints, liver and haematopoietic systems, with clinical manifestations of fatigue or decreased endurance at the outset [7, 41, 42]. These often present before GVHD-BOS diagnosis [7]. Overall, the diagnostic features of GVHD-BOS are similar to those of CLAD, with primary pulmonary function impairment [41].
The US National Institutes of Health definition is shown in table 1 [41]. These features include FEV1/vital capacity (forced or slow, whichever is greater) <0.7 or the 5th percentile of predicted and FEV1 <75% of predicted and with ≥10% decline over <2 years in the absence of an infective cause, and evidence of air trapping or other signs of BOS (small airway thickening or bronchiectasis) on CT or evidence of gas trapping on lung function testing.
Unlike CLAD, specific GVHD-BOS phenotypes have not been defined [43]. However, Bergeron et al. [44] proposed two different lung function patterns in patients with GVHD-BOS. The first is a typical obstructive defect characterised by FEV1/FVC ratio <0.7, and the second is atypical with FEV1 <80% and FVC <80% but normal lung capacity, such that the FEV1/FVC ratio was >0.7. Outcomes did not differ between the two groups, but patients with the typical pattern had fewer centrilobular nodules on CT [44]. These data need to be further explored as they could also represent a restrictive GVHD lung disease or a subset of patients with BOS who do not demonstrate obstruction due to extrapulmonary constraints (e.g. sclerotic GVHD of the respiratory girdle).
The trajectory of FEV1 decline in patients with GVHD-BOS is heterogeneous and deterioration may happen rapidly [45, 46]. Studies have shown that patients with a rapid decline in lung function (25% FEV1 decline) during the first 3 months of GVHD-BOS, those with poorer FVC at diagnosis of GVHD-BOS or those with nontuberculous mycobacteria in bronchoalveolar lavage (BAL) culture have worse survival than those who do not [14, 45–47].
Pathophysiology and aetiology
CLAD
The temporal relationship between the development of CLAD and infectious diseases (e.g. chronic pulmonary P. aeruginosa and CARV) supports the concept that one pathway of CLAD is a microbe–allograft–host interaction, whereby the infectious pathogen causes allograft cells to release chemokines that recruit host leukocytes to the site that then recognise the airways as non-self [48, 49]. Other pathways involve endogenous molecules released by injury to small airways or their microvasculature following diverse injuries including ACR [50]. The influx of leukocytes precipitates an allo-response causing graft dysfunction [48, 49]. Thus, CLAD is a host-versus-graft (rather than a graft-versus-host) disease. The inescapable conclusion is that both are immune-regulated phenomena, which may explain why BOS is common after lung transplantation and HSCT.
Dysfunction of the regulatory mechanisms and induction of an acute inflammatory response generate a positive feedback loop and amplify the immune response, causing the pathological process to transition from an acute to a chronic response [50]. Humoral (adaptive) immune activation may be a contributing mechanism determining CLAD-RAS phenotype development [51]. Continuous exposure of the airway epithelium to inflammatory processes can lead to fibroblast recruitment and eventually extracellular matrix remodelling [25]. Acute fibrinous and organising pneumonia appears to be an early event in this process [25, 52].
The histological features of CLAD can be heterogeneous. The dominant and most frequent finding is obliterative bronchiolitis. Other common findings include variable grades of ACR including perivascular and/or peribronchiolar lymphocytic infiltrates, pleuro-parenchymal fibro-elastosis, and microvascular damage [53].
GVHD-BOS
The early processes leading to BOS in GVHD differ from those in CLAD, but the eventual histological changes are relatively similar. As described earlier, BOS after HSCT is a manifestation of chronic GVHD. While less is known about the aetiology of BOS after HSCT, chronic GVHD is caused by central tolerance failure and B-cell and auto-antibody production [40, 54]. T-cells play a major role in the initiation of GVHD; the subset of T-cells that are primarily responsible for the development of pulmonary GVHD are not characterised, although CD4+ T-helper 17 cells are likely involved [40].
The pathology of GVHD-BOS is less well defined than BOS after lung transplantation due to a lower volume of surgical lung biopsies and lower autopsy rates. One study showed two distinct patterns: constrictive bronchiolitis obliterans (CBO) and lymphocytic bronchiolitis [55]. CBO demonstrates marked bronchiolar narrowing with fibrous lesions and hyperplasia of the epithelium, whereas in lymphocytic bronchiolitis, fibrosis is absent, and there is bronchiolar dilatation, and epithelial thinning, necrosis or disappearance [56]. Patients with lymphocytic bronchiolitis tend to have better survival than those with CBO [55].
The pattern of morphological changes on pulmonary micro-CT is similar in patients with obstructive CLAD and those with GVHD-BOS [57], with both having a reduced number of terminal bronchioles [57].
Diagnosis
Early diagnosis of CLAD may be more likely than GVHD-BOS, since pulmonary function is monitored more frequently in lung transplant recipients. In contrast, the major concern after HSCT is haematological malignancy relapse, hence pulmonary function testing is generally less frequent and sporadic [45]. As a result, many HSCT patients potentially miss the opportunity for early intervention for BOS [45].
Current guidelines recommend implementing early investigations for CLAD as soon as the condition is suspected (i.e. ≥10% reduction in baseline FEV1) [3]. Spirometry is a key monitoring and diagnostic tool in both lung transplant or HSCT recipients with suspected pulmonary complications, but extrapulmonary manifestations may affect lung function tests in GVHD-BOS patients. Home spirometry may detect early pulmonary function decline but is not a substitute for office testing [58] and requires periodical calibration of the home spirometer against the laboratory spirometer and assessment of the patient's technique. Home spirometry may be a useful way to limit clinic visits (e.g. during the SARS-CoV-2 pandemic).
Once lung function decline is confirmed, possible causes should be investigated using bronchoscopy with visual airway inspection, transbronchial biopsies where indicated, BAL, TLC testing (e.g. plethysmography) and CT imaging (supplementary table S2) [3]. Lung biopsy (usually by the transbronchial route) is considered the gold standard diagnostic modality to rule out other causes of FEV1 decline, such as ACR. However, the risks of biopsy (bleeding and pneumothorax) may outweigh the benefits in some patients, particularly those with GVHD-BOS, in whom noninvasive methods play an important role in diagnosis [3, 59–61].
Small airway brushings may detect a lymphocytic gene expression signature in patients with CLAD, which may not be apparent in transbronchial biopsies [62]. Functional magnetic resonance imaging (MRI) can assess regional changes in lung function assisting early CLAD detection [31] but is expensive and not universally available [59].
CT or CXR are commonly used to identify opacities, but CT is preferred due to greater sensitivity and specificity than CXR and better visualisation of changes in lung parenchyma and small airways. The presence of air trapping, and especially its increase, as seen on high-resolution chest CT during inspiration and expiration is supportive of BOS diagnosis [63, 64]. Radiation exposure is higher with CT than conventional CXR but can be substantially reduced by low-radiation protocols. While the initial use of CXR followed by CT in patients with suspicious CXR findings may be optimal, air trapping can limit the utility of CXR in GVHD-BOS patients [59].
Typical chest CT findings in CLAD (RAS/mixed phenotype) are opacities (ground glass appearance, consolidation, small linear or reticular densities) and/or increased pleural thickening indicative of fibrosis [3]. Centrilobular opacities, air trapping and bronchial wall thickening may be present on CT of patients with BOS after lung transplantation or GVHD, but pleural thickening is absent or rare after GVHD [64].
An emerging modality in BOS diagnosis and assessment is parametric response mapping (PRM), in which expiratory and inspiratory CT scans undergo voxel-based analysis [65, 66]. In patients with BOS after lung transplantation, PRM correlates with FEV1 decline (at least in patients without a restrictive pattern), whereas air trapping does not [66]. In patients with BOS after HSCT, PRM can detect BOS even in those with concurrent infection [65]. PRM is useful for monitoring BOS progression after either type of transplant [65, 66], but further data are required.
MRI has been investigated for morphological assessment of transplanted lungs, and some MRI parameters may be early markers of CLAD [67, 68], but further data are needed before MRI is routinely used for BOS assessment.
Perhaps the most innovative strategy to monitor ACR and antibody-mediated rejection revolves around the understanding that these responses cause cell death and the release of donor-derived cell-free DNA into the circulation, which may be used as a noninvasive, quantitative marker to track the events leading to CLAD [69].
Artificial intelligence (AI) is another innovation that can be applied to understanding rare diseases. One example is the identification of lung GVHD after HSCT by quantitative imaging [70]. AI has the potential to increase our understanding of the similarities and differences between CLAD-BOS and GVHD-BOS.
Current treatment approaches
CLAD-BOS
Initially, any precipitating or underlying conditions should be identified and treated, including ACR, infection, lymphocytic bronchiolitis, GERD or others [3], and the patient's maintenance immunosuppressive regimen should be optimised (figure 1a). A consecutive case series indicated that early management of BOS, when it is less severe, may be efficacious in stabilising declining lung function parameters at a higher plateau. Based on their years of clinical practice experience, the authors agreed that early introduction of therapies may mitigate progression to more severe BOS [71]. If ACR is diagnosed, it should be treated as per standard protocols avoiding prolonged courses of high-dose steroids [3, 24]. There is evidence suggesting that the prevalence of BOS after lung transplantation is lower in patients receiving tacrolimus versus cyclosporine [72] and that switching from cyclosporine to tacrolimus stabilises lung function [24, 73].

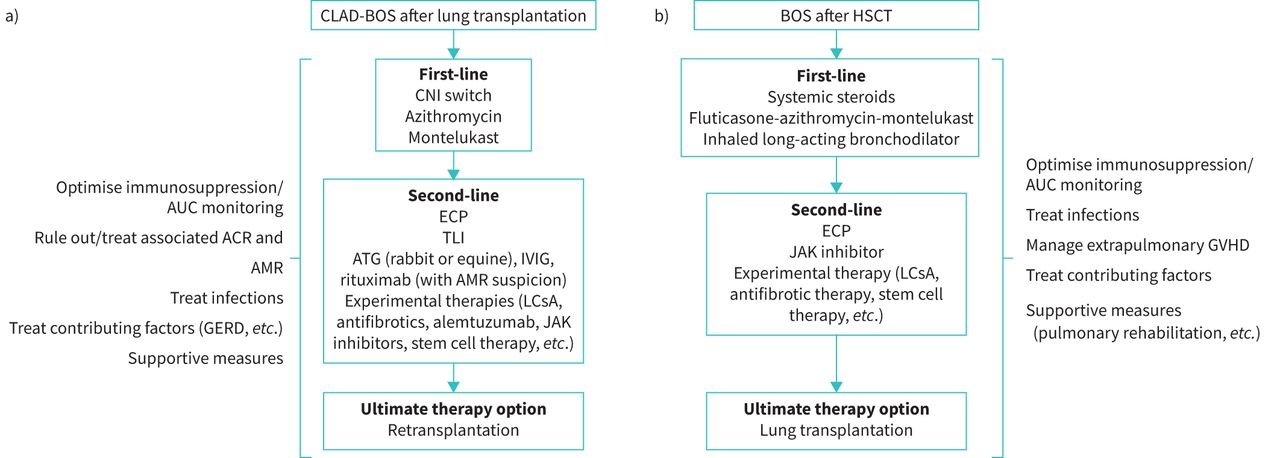{kind=link}
Treatment approach for BOS after a) lung transplantation and b) HSCT. ACR: acute cellular rejection; AMR: antibody-mediated rejection; ATG: antithymocyte globulin; AUC: area under the plasma concentration–time curve; BOS: bronchiolitis obliterans; CNI: calcineurin inhibitor; CLAD: chronic lung allograft dysfunction; ECP: extracorporeal photopheresis; GERD: gastro-oesophageal reflux disease; GVHD: graft-versus-host disease; HSCT: haematopoietic stem cell transplant; IVIG: intravenous immunoglobulin; JAK: Janus kinase; LCsA: aerosolised liposomal cyclosporine; TLI: total lymphoid irradiation.
Among available treatments, the most evidence exists for azithromycin [74–81], including data from a randomised, placebo-controlled trial [82]. In observational studies, azithromycin was associated with a FEV1 increase of 16–18% predicted [74, 75, 78], and an absolute FEV1 increase of 0.11–0.86 L [74, 78–80]. However, response rates were only 29–50% in these studies, indicating that ≥50% of patients showed no improvement [74, 75, 78–81]. Factors associated with a greater likelihood of response were airway neutrophilia (detected by BAL) [79, 81] and early treatment initiation [76]. In the randomised study, nine out of 23 patients on azithromycin (39%), but none of those in the placebo group, responded to treatment with a FEV1 increase of ≥10% predicted (p=0.002). The difference in FEV1 between the azithromycin and placebo groups was 0.278 L (p<0.001) [82]. Where safety data were reported, azithromycin was generally well tolerated [75, 82]. One patient developed laryngeal oedema (serious) that resolved after azithromycin discontinuation [75]. The most frequent adverse effects were gastrointestinal disorders. The recent ISHLT consensus states that azithromycin should be initiated as early as possible, even before any definite BOS diagnosis has been made [3], based on its effects on lung function [76, 77, 81]. The optimal dosage and duration of azithromycin have not been established [78, 80, 81]. BOS may occur despite long-term maintenance azithromycin.
The leukotriene antagonist montelukast has also been investigated for BOS treatment [83–85]. Montelukast is an oral treatment for persistent asthma [86], showing antifibrotic effects in animal models of BOS after lung transplantation [87]. In a pilot study of 11 patients with low neutrophilia on BAL (<15%) who were expected to be poor responders to azithromycin, montelukast slowed FEV1 decline [85]. Similarly, attenuation of FEV1 decline was seen in a retrospective study of 153 BOS patients, which also showed significantly longer survival in patients with response to montelukast than in those who did not respond [83]. However, a randomised, placebo-controlled trial failed to demonstrate any effect of montelukast on FEV1 or graft survival in BOS patients, although benefit was seen in patients with early-stage BOS [84]. As this study only included 15 patients in each treatment arm, further research in larger patient cohorts is warranted.
Second-line options include extracorporeal photopheresis (ECP) or total lymphoid irradiation (TLI) [3]. ECP slows the rate of FEV1 decline in patients with CLAD-BOS [88] and is probably more effective in patients with the BOS than the RAS phenotype [89]. Some data support an improvement in survival among patients with CLAD receiving ECP, but overall, the evidence supporting ECP in BOS is considered to be Class IIB, level C [90]. Moreover, ECP is expensive, not universally available and potentially burdensome for some patients. It requires secure venous access and multiple treatment sessions initially over a short period [90]. On the other hand, ECP is generally well tolerated, with no major adverse effects reported in a large patient series [90].
TLI has been shown to slow the rate of FEV1 decline in BOS patients, including those not responding to azithromycin [91, 92], but studies to date have been small and observational [90]. In addition, not all patients are able to complete the required number of treatment sessions due to bone marrow suppression or infection [91].
Re-transplantation remains an option in carefully selected patients with BOS who fail available first- and second-line treatment options.
GVHD-BOS
Other manifestations of chronic GVHD or non-infectious complications have been described, including interstitial lung diseases, but specific management of these conditions differs from the approach to BOS [7].
The first step in managing BOS after HSCT is to ensure that comorbidities and potential precipitating factors (e.g. GERD) are managed, and that immunosuppressive treatment is optimised [60]. Treatment of BOS is often additive to that targeting the extrapulmonary manifestations of GVHD; therefore, clinicians should manage the immunosuppression as a whole (figure 1b). Long-term corticosteroids are not considered beneficial for BOS after HSCT but may be part of therapy for GVHD.
Currently, there is limited evidence to guide treatment of BOS after HSCT [93]. The European Society for Blood and Marrow Transplantation recommends the combination of fluticasone, azithromycin and montelukast (FAM), with a steroid pulse and rapid taper over 1 month (class 2A evidence) [93]. This recommendation was based on data from a non-randomised study in 36 patients with BOS after HSCT who received FAM plus a steroid burst (planned to be 4 weeks total of prednisone commencing at 1 mg·kg−1·day−1 and initiating taper after 1 week) [94]. Notably, most patients were on standard treatment for chronic GVHD (i.e. sirolimus or a calcineurin inhibitor), which likely contributed to BOS control. After 3 months, only 6% of patients met the definition of treatment failure (FEV1 decline of ≥10%) and 17% met the definition at 6 months [94]. Patient-reported outcomes showed significant improvements from baseline at 3 months, including in social functioning, mental and emotional well-being, the 6-min walk test and symptom severity. FAM was generally well tolerated, and only one patient discontinued treatment because of adverse events [94].
The data for azithromycin alone are less robust [95]. While one study suggested that azithromycin alone as a preventive strategy for BOS may be linked to relapse, more recent larger studies did not show increased risk of relapse with azithromycin post-GVHD, although increased risk of secondary cancers with higher use of concomitant steroids was observed [96, 97]. A combination of inhaled long-acting bronchodilator plus inhaled corticosteroid has been shown to improve FEV1 at 1 month (by 200 mL and 12%) in newly diagnosed patients with mild-to-moderate BOS, without the use of systemic corticosteroids [98]. Patients may also benefit from supportive care including prophylaxis for infection, pulmonary rehabilitation, nutritional support and treatment for GERD [60, 99].
There are no standard second-line treatment approaches to lung chronic GVHD, so these are determined by national, local or institutional guidelines [93]. Some retrospective data support the use of ECP, with ECP improving survival in HSCT with BOS without significantly impacting pulmonary function [100]. Given the overall paucity of data, enrolment in a clinical trial should be prioritised as a second-line treatment approach [60, 93].
In the USA, other agents for chronic GVHD are being explored: the Bruton's tyrosine kinase inhibitor ibrutinib is approved for the second-line treatment of chronic GVHD [101], as is the Janus kinase (JAK) inhibitor ruxolitinib, but there are no data regarding their efficacy for BOS [102]. One trial evaluated the use of a tumour necrosis factor-α inhibitor, etanercept, and showed benefit (i.e. ≥10% improvement in absolute FEV1 or FVC) in one-third of patients with BOS [103].
As with CLAD, the ultimate treatment option is a lung transplant [104].
Future directions
Hypothesis-driven novel approaches
Any novel therapeutic approach for BOS management is driven by the understanding of the disease pathogenesis of obliterative bronchiolitis. In this regard, the immunogenicity of the allograft is considered to be important, as obliterative bronchiolitis develops due to an injury–response mechanism, where the small airways are targeted as non-self after upregulation of HLA on respiratory epithelial cells [105]. Early studies showed that bronchiolar epithelial cells from lobectomy samples of former smokers expressed both Class 1 and Class 2 HLAs, as did small airways in explants from patients undergoing re-transplantation for obliterative bronchiolitis [106, 107]. Despite this promising groundwork, the potential role of HLA presentation on small airway epithelia has only recently been reconsidered with the acceptance of the relevance of antibody-mediated rejection as a cause of allograft injury [108]. Indeed, the presence of donor-specific antibodies against Class 2 HLAs, especially those against HLA-DQ2, is considered a major risk factor for CLAD development [109].
Ischaemia plays an important role in scar formation after injury, and damage to the “watershed” microvasculature of the terminal bronchioles appears to compound the geometric factors which determine why small airways bear the brunt of the rejection response [110]. Obliterative bronchiolitis lesions seen on micro-CT scans are essentially scars at focal segments of small airways where the remnants of the external elastic lamina are best visualised on histopathology by elastin van Gieson staining [4]. While the upstream bronchiole may appear normal or simply show thickening of the basement membrane, the acinus subtended by the affected bronchiole is excluded from gas exchange [4]. Evidence for an immunological aetiology for BOS is supported by a retrospective analysis of transbronchial lung biopsies, where the severity of lymphocytic bronchiolitis was strongly correlated with the time to develop BOS [111].
Understanding the fundamental pathogenesis of BOS and applying this knowledge in clinical practice is essential to mitigate the risk of BOS after lung transplantation or HSCT. Since HLA presentation is thought to be the main target, efforts should be directed towards effective prevention of trigger events which upregulate antigen presentation and damage small airways. Trigger events probably include T-cell-mediated ACR, CARV infections with lower respiratory tract tropism (such as respiratory syncytial virus), certain coronavirus infections, bacterial infections (such as P. aeruginosa, mycoplasma and chlamydia) and inflammation from aspiration of gastric contents [112]. The possibility of autoimmunity to cryptic self-antigens liberated by these triggers is supported by the presence of self-antigens in microvesicles and the development of autoantibodies against Type V collagen and K-α1 tubulin [113, 114].
Novel therapies will need to embrace these concepts to successfully prevent tissue damage and modify aberrant repair. A multifaceted approach that includes risk factor management for the triggers outlined above is essential, starting with a global response to ameliorate the toxic effects of air pollution on small airways and protect airways from direct/indirect exposure to cigarette smoke and e-cigarette emissions [115].
In line with these hypotheses, several agents have been or are being investigated for the treatment of BOS after lung transplantation or HSCT, as discussed below.
Aerosolised liposomal cyclosporine
Aerosolised cyclosporine delivers immunosuppressive therapy directly to the allograft, where it acts locally at the immune activation site, limiting systemic exposure [116]. A liposomal cyclosporine formulation for aerosolised delivery (Zambon), currently undergoing phase III development, has been granted orphan drug status by the US Food and Drug Administration [117]. This formulation showed good lung deposition after nebuliser administration in lung transplant recipients [116]. In a randomised study, adding aerosolised liposomal cyclosporine to standard care improved or stabilised a range of lung function parameters and was significantly more effective than standard care alone [118], but this study was open-label without placebo control. Treatment was well tolerated, with no increase in serious adverse events with aerosolised liposomal cyclosporine versus standard care alone; adverse events included conjunctivitis, pharyngitis and productive cough [118].
The Boston clinical trial programme is investigating aerosolised liposomal cyclosporine in patients with BOS after lung transplantation or HSCT (table 2); results will become available over the next 2–3 years.
Clinical trials with aerosolised liposomal cyclosporine
JAK inhibitors
JAK inhibitors are used for the treatment of solid tumours and are in development for autoimmune conditions [119]. Ruxolitinib (Incyte, Novartis, Wilmington, DE, USA) is approved for the use of steroid-refractory GVHD in the USA [102] and is the most studied agent from this class for the treatment of BOS after HSCT or bone marrow transplantation. To date, published data are limited to small case series or case studies in adults or children with steroid-resistant BOS [120–123]. However, some studies have suggested a potential role for these agents in the treatment of BOS [120–123]. An open-label phase II study (NCT03674047) is underway in the USA in patients with newly diagnosed or established BOS after HSCT (completion expected in March 2023).
B-cell-directed therapies
B-cells are important mediators of chronic GVHD [124, 125] and are implicated in the development of CLAD [126]; thus, treatments inhibiting B-cell activation may be useful for BOS treatment after lung transplantation or HSCT. However, B-cell-directed therapies tend to be associated with significant safety concerns, including cytopenias, immunosuppression and infections [127, 128].
Rituximab is a chimeric human/mouse monoclonal antibody against CD20 that has cytolytic activity against B-cells and is approved for the treatment of B-cell lymphomas [127]. Small case series suggest that rituximab may be effective in improving lung function in patients with BOS after lung transplantation with concurrent antibody-mediated rejection [129–131]. There are less data regarding its use in patients with BOS after HSCT. One report of three cases described lung function stabilisation in only one patient, but the authors acknowledged that rituximab was only initiated in severely ill patients, and its efficacy if used at an earlier stage of BOS could not be precluded [131].
Alemtuzumab is an anti-CD52 monoclonal antibody approved for use in B-cell lymphomas [128]. A database analysis suggested that using alemtuzumab as part of the induction regimen reduced the 5-year risk of BOS after lung transplantation compared with basiliximab-based induction or no induction [132]. Two retrospective studies and a case series (n=10) reported slowing or reversal of lung function decline with alemtuzumab in a high proportion of patients with BOS after lung transplantation [133–135]. Alemtuzumab appeared to be similarly effective to ECP [134]. Patients with early-stage BOS were more likely to respond to alemtuzumab than those with late-stage BOS [132]. Notably, this strategy would mitigate anticancer and anti-infection control in patients with BOS after HSCT and would thus be associated with higher risk.
Antifibrotic treatments
The antifibrotic agents pirfenidone and nintedanib are used in the treatment of chronic fibrosing pulmonary conditions, such as idiopathic pulmonary fibrosis [136, 137], leading to speculation that they may ameliorate the fibrotic changes in BOS after transplantation. Preclinical studies demonstrated antifibrotic effects with pirfenidone in animal models of BOS or post-transplant pulmonary complications [138–141]. Preliminary data from a case series of 11 RAS patients showed that pirfenidone stabilised lung function during long-term treatment and provided a bridge to a second lung transplant in three patients (27%) [142]. Published data with nintedanib are limited to case reports: one showed a clinical benefit in a patient with BOS after HSCT [143], and another showed no benefit in a patient with BOS after lung transplantation [144].
Clinical trials with both agents are underway in post-transplant patients with BOS (table 3). The results of the EPOS trial (pirfenidone versus placebo) were negative [145]. Another small phase II study (PIRCLAD; NCT03359863) is also investigating pirfenidone in patients with RAS (n=10), with completion expected in October 2021. A potential role for mesenchymal stem cell therapy is being explored in a randomised controlled study in Australia, but results are not yet available.
Clinical trials with antifibrotic treatments
Conclusions
Management strategies for BOS after lung transplantation or HSCT are similar, although a key difference is the need to manage systemic GVHD in HSCT patients. Currently, treatment options for either condition are limited and novel treatments are urgently needed, particularly treatments which not only slow or stabilise disease progression but also potentially reverse the pathological changes and thereby provide clinically meaningful survival and quality of life outcomes.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00185-2022.SUPPLEMENT
Acknowledgements
We would like to thank Catherine Rees of Springer Healthcare Communications who wrote the outline and first draft of this manuscript. This medical writing assistance was funded by Zambon.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: A.R. Glanville contributed to the concept and design of the manuscript, chaired the panel discussion, and manuscript drafting and approval. All authors contributed to the panel discussion and manuscript review.
Conflict of interest: A.R. Glanville has received an honorarium for an advisory role from Zambon and is chair of the Zambon DSMB for the Boston trials.
Conflict of interest: G.M. Verleden has received an honorarium for an advisory role from Zambon.
Conflict of interest: M. Perch has received an honorarium for an advisory role from Zambon, a research grant (institutional) from Roche, speaker fees from Novartis, GSK and Therakos, and other financial support from Boehringer.
Conflict of interest: E.D. Lease has received an honorarium for an advisory role from Zambon.
Conflict of interest: G-S. Cheng has received an honorarium for an advisory role from Zambon.
Conflict of interest: A. Bergeron has received an honorarium for an advisory role from Zambon.
Conflict of interest: C. Benden has received an honorarium for an advisory role from Zambon and speaker fees from Therakos.
Conflict of interest: J. Gottlieb has received an honorarium for advisory roles from Zambon, research grants from Zambon and Deutsche Forschungsgemeinschaft, and advisory funding from Theravance, Merck and Altara.
Conflict of interest: J.L. Todd has received an honorarium for an advisory role from Zambon, Altavant and Natera, and research grants (institutional) from Boehringer Ingelheim, AstraZeneca and CareDx.
Conflict of interest: K.M. Williams has received an honorarium for an advisory role from Zambon.
Support statement: Zambon provided financial support for the expert panel meeting and manuscript development. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 13, 2022.
- Accepted May 18, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










