





Abstract
Introduction Pulmonary vascular remodelling in chronic obstructive pulmonary disease (COPD) has detrimental consequences for lung physiology. The aim of our study was to provide a comprehensive size-based morphometric quantification of pulmonary arterial remodelling in smokers and in patients with small airway disease (SAD) or COPD.
Method Movat's pentachrome staining was performed on lung resections for 46 subjects: 12 never-smoker normal controls (NC), six normal lung function smokers (NLFS), nine patients with SAD, nine patients with mild-to-moderate COPD who were current smokers (COPD-CS) and 10 patients with mild-to-moderate COPD who were ex-smokers (COPD-ES). Following a size-based classification of pulmonary arteries, image analysis software was used to measure their number, total wall thickness, individual layer thickness and elastin percentage.
Results All pathological groups showed decreased numbers of pulmonary arteries compared with the NC group in all artery sizes. Arterial wall thickness was greater in NLFS and COPD-CS than in NC. Thickness in COPD-ES was decreased compared with COPD-CS. Intimal thickness was greater in all pathological groups in all arterial sizes than in the NC group. Medial thickness was also greater in small and medium arteries. Intimal thickness of larger arteries in COPD-CS correlated negatively to forced expiratory volume in 1 s/forced vital capacity (FVC) % and forced expiratory flow at 25–75% of FVC. Elastin deposition in small arteries was greatest in COPD-CS. Intimal elastin deposition had a more negative correlation with intimal thickness in NLFS and SAD than in COPD-CS.
Conclusion Smoking, SAD and mild-to-moderate COPD are associated with pruning and a decrease in the number of pulmonary arteries, increased wall thickness and variable elastin deposition. These changes were associated with worse airway obstruction.
Abstract
Pulmonary arterial remodelling occurs in smokers and in patients with small airway disease or COPD, where it affects lung function and may exaggerate pulmonary hypertension. Endothelial-to-mesenchymal transition might be central to these vascular changes. https://bit.ly/3QF5heH
Introduction
Chronic obstructive pulmonary disease (COPD) is a major cause of mortality worldwide [1]. The disease is characterised by progressive airflow limitation and is associated with a variable chronic inflammatory response to cigarette smoking and environmental pollutants, especially gases emanating from fossil fuel burning [2, 3]. As a result, COPD patients develop several structural changes in the lung, including small airway wall thickening and narrowing, large airway squamous cell metaplasia, mucus hypersecretion and smooth muscle hyperplasia [4–7]. Further manifestations can include hyperinflation, hypoxaemia, hypercapnia and increases in pulmonary vascular pressures [1], with the latter resulting from vascular remodelling starting during the early disease stages [8, 9]. Vascular modifications can lead to pulmonary hypertension (PH), a common comorbidity associated with COPD. PH is associated with increased risk of exacerbation [10]. Moreover, PH can cause right ventricular failure and can affect survival rates [10].
Vascular remodelling is not only restricted to patients with established COPD but is also seen in smokers with normal lung function [11]. The most prominent feature of vascular remodelling in these groups of patients is intimal hyperplasia, primarily due to endothelial and smooth muscle cell proliferation [12]. Narrowing of arteries due to luminal encroachment and deposition of elastin and collagen are other vascular changes reported in COPD patients [13, 14]. Computed tomography studies have reported a decreasing number of pulmonary vessels along with COPD progression [15] and pruning of small arteries in smoking-related COPD [16]. However, the relative contribution of cigarette smoke and COPD-induced hypoxia in the remodelling of the pulmonary vasculature in different cohorts of smokers and COPD patients remains undetermined [17]. Furthermore, the magnitude of remodelling in the individual layers of the arterial wall, their extracellular matrix composition and consequences for lung function are also currently unavailable in the literature.
The pathogenesis of PH in smokers and in patients with COPD remains undefined, and is currently classified under the broad umbrella term of pulmonary vascular changes [18]. Although PH has been diagnosed in patients with late-stage COPD, changes that lead to PH could commence early in the disease, with structural changes in vessels primarily due to endothelial dysfunction [17]. The trigger for such vascular changes is likely a reaction to toxic chemicals associated with cigarette smoking [17]. Chronic hypoxia resulting from progressive airflow limitation and parenchymal destruction are other important factors for vascular remodelling [1]. Given the significant role of the pulmonary vasculature in gas exchange and its maintenance of lung homeostasis, its dysfunction during pathological conditions will critically impact lung physiology and disease progression.
Few investigators have published detailed histopathological analyses of the pulmonary vasculature in well-characterised smokers and in patients with COPD [14, 19]. In this study we compared pulmonary arterial numbers and remodelling in healthy never-smoker normal controls (NC) with smokers with normal lung function (NLFS), patients with small airway disease (SAD) and patients with mild-to-moderate COPD who were current smokers (COPD-CS) or ex-smokers (COPD-ES). We provide quantitative assessments of the total wall thickness, the thickness of individual layers and the presence of elastosis and study their relation to lung physiological parameters.
Material and methods
Study population
This study was approved by the Tasmania Health & Medical Human Research Ethics Committee (Ethics no. H0012374). Surgically resected human lung tissues were available from our biobank. Tissue was used from participants with primary non-small cell lung cancer who consented to research procedures. The resected material was taken far from the primary tumour and contained non-cancer-affected pulmonary blood vessels. A single block and tissue section were used for each participant. Participants were grouped into NLFS (n=6); SAD (n=9), with SAD defined as forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) ratio >70% and expiratory limb scalloping and forced expiratory flow at 25–75% of FVC (FEF25–75%) <68% predicted [20]; and COPD-CS (n=9) and COPD-ES (n=10), with COPD categorised as Global Initiative for Chronic Obstructive Lung Disease (GOLD) 1 or 2 (FEV1/FVC <70% and FEV1 predicted value >50%). Participants classed as COPD-ES had at least 6 months of smoking-free history. In addition, tissue from 12 subjects who died of causes other than respiratory diseases was obtained from the James Hogg Lung Registry, University of British Columbia (ethical approval from the Providence Health Care Research Ethics Board H00–50110) to use as the NC group. Subject demographic and lung function measures, obtained from participants as part of standard clinical practice timed closely to the resection of lung tissue, are provided in table 1.
Participant demographics
Movat's pentachrome staining
Movat's pentachrome staining was performed in tissue sections from all groups following our previously published work [21].
Pulmonary arterial count and classification
Tissue sections were imaged with a ×4 objective in a vertical uni-direction, avoiding image duplication using a Leica IC50W digital camera connected to Leica-DM 500 microscope. Image analysis was performed using Image-Pro Plus 7.0. Pulmonary arteries were distinguished based on their nearly rounded structure, thickness, and defined internal and external elastin layers, whereas veins were elongated, thinner and had only a single elastin layer. First, maximum external diameter (end to end of adventitia) and luminal diameter (end to end of inner intima) were measured (figure 1a). Next, arteries with a maximum diameter between 100 µm and 1000 µm were segregated into three groups, with arteries measuring 100–299 µm classed as small, 300–499 µm as medium and 500–999 µm as large (figure 1b) [14, 21].

Methods for measurement of a) external and luminal diameter and b) categorisation based on external length. c) Strategy for the selection of an individual layer in the arterial wall.
Arterial number normalised to the parenchymal tissue density
We obtained non-overlapping images from the tissue section of each subject, consisting of arterial range 100–1000 µm. Arterial counts for all ranges along with the parenchymal tissue density calculations for each tissue section were done using a similar strategy as previously reported [21]. In brief, the absolute arterial numbers were first estimated for each tissue section by manual counting. Then the parenchymal tissue density was calculated by selecting the entire parenchymal tissue area using an automated selection program (Image-Pro Plus v7 software). The sum of the parenchymal tissue density for each subject was calculated. The total arterial number counts were normalised to total parenchymal tissue density for all subjects.
Pulmonary arterial thickness measurement
The total arterial wall thickness was calculated by subtracting the luminal diameter from the maximum diameter (figure 1a). For the differential assessment of all three arterial wall layers, non-overlapping images of all the previously captured and categorised arteries were magnified further depending on the arterial size. Smaller arteries with an external diameter of 100–199 μm were captured using ×63 magnification, arteries of 200–599 μm were captured using ×40 magnification and larger arteries of 600–999 μm were captured using ×20 magnification. All the categorised arteries with a clear distinction of three different layers were captured for each arterial range of every individual subject. An online random number generator, stattrek, randomly selected five images per subject in each arterial size range.
Measurements for each layer were carried out as follows. The intima thickness was measured from the outer luminal to the inner elastin layer. Media layer thickness was measured from the external layer of the inner elastin membrane to the internal lining of the external media's elastin membrane. Adventitial thickness was measured from the media's external elastin layer to the artery's outermost connective tissue. Lines were drawn manually to predefine these layers (figure 1c). Based on the orientation of the arteries, a horizontal, vertical or curved distance was calculated using the measurement tools of the imaging software.
Pulmonary arterial elastin measurement
A similar strategy to thickness measurement was followed for image acquisition and randomisation for arterial elastin measurement. The method used for individual layer thickness measurement was used to select the area of interest for the individual layer elastin deposition (figure 1c). To quantify elastin, the blue/black colour that elastin shows on the image was manually selected and counted for the area of interest, and then the total dark object in the same area was also measured. Finally, the sum elastin percentage was calculated using the following formula:
Statistical analysis
Cross-sectional data were tested for a normal distribution using the D'Agostino–Pearson omnibus normality test. Next, two-way ANOVA was performed with mean intergroup comparisons done using Fisher's least significant difference test. Based on the ANOVA result, univariate Spearman or Pearson r was used for correlation analysis. All analyses were done using GraphPad Prism 9.3, with p≤0.05 considered significant.
Results
Morphological assessment of pulmonary arteries
Microscopic evaluation of the stained lung resections from all smoking groups (NLFS, SAD, COPD-CS and COPD-ES) showed several structural modifications in the arteries and lung parenchyma compared with the NC group. The alveolar septum and the arterial wall were thicker in all four smoker groups than in the NC group. Other structural changes, such as luminal encroachment by the proliferative intimal cells and muscular hypertrophy within the medial layer, were noted in the four smoking groups but not in the NC group. While the inner and outer elastin lamina remained intact in the NC group, they were disorganised or diffused across the increasingly thickened arteries in the COPD groups (figure 2).

Representative images of Movat's pentachrome-stained pulmonary arteries (a–o) and parenchymal area (×4 magnification) (p–t) for never-smoker normal controls (NC), normal lung function smokers (NLFS), patients with small airway disease (SAD), current smokers with chronic obstructive pulmonary disease (COPD-CS) and ex-smokers with chronic obstructive pulmonary disease (COPD-ES). Arteries grouped by size as a–e) 100–300 µm (×20 magnification), f–j) 300–500 µm (×20 magnification) and k–o) 500–1000 µm (×10 magnification). In p–t, increased parenchymal tissue density and intimal thickening with luminal narrowing can be seen in the four pathological groups.
Parenchymal tissue density and arterial number
In our assessment of the airway alveolar region, the parenchymal tissue density was greater in all four smoking groups than in the NC group (figure 3a). Total arterial counts were smaller in the four smoking groups than in the NC group (figure 3b). Similar trends were seen across the arterial sizes: the number of small (100–300 µm), medium (300–500 µm) and large (500–1000 µm) arteries was smaller in the smoking groups than in the NC group (figure 3c).

Comparison of a) parenchymal tissue density, b) total number of arteries normalised to parenchymal tissue density and c) number of arteries normalised to parenchymal tissue density in arterial ranges of 100–300 µm, 300–500 µm and 500–1000 µm. NC: never-smoker normal controls; NLFS: normal lung function smokers; SAD: patients with small airway disease; COPD-CS: current smokers with chronic obstructive pulmonary disease; COPD-ES: ex-smokers with chronic obstructive pulmonary disease. *: p≤0.05; **: p≤0.01; ****: p≤0.0001.
Pulmonary arterial wall thickness
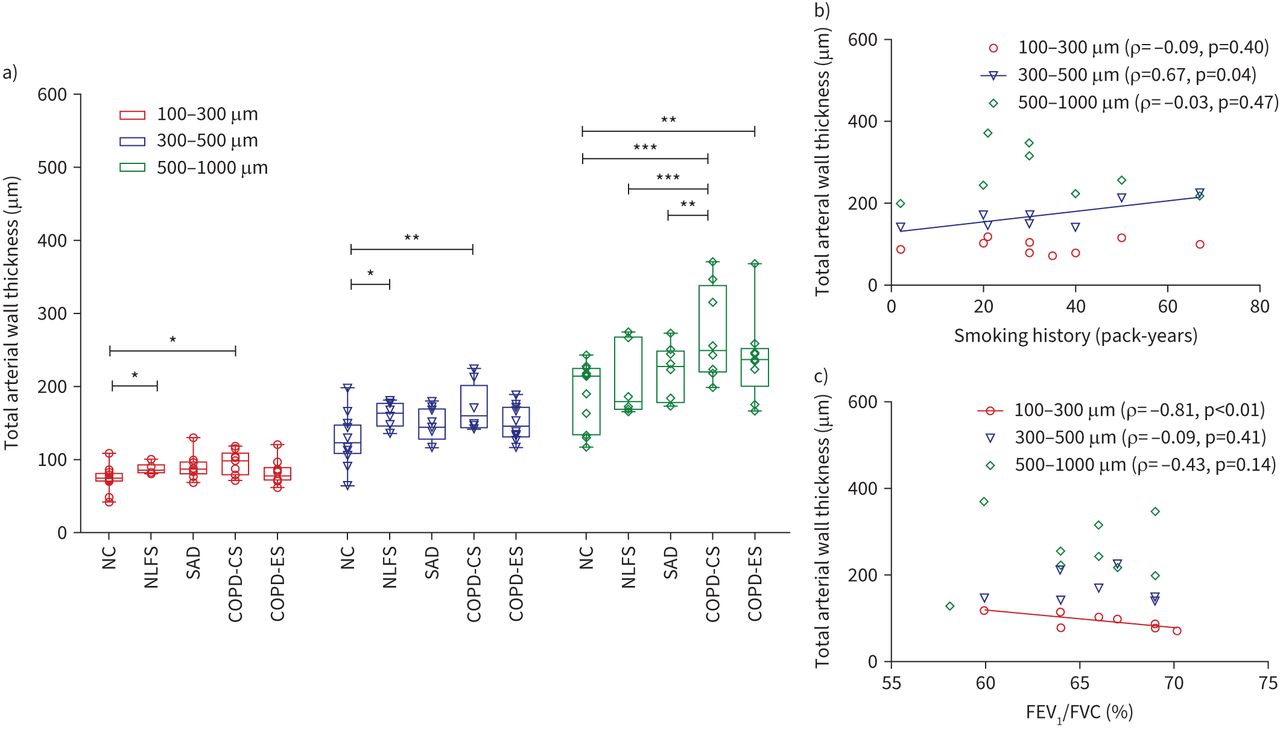
The NLFS group and COPD-CS group had thicker walls in small and medium-sized arteries than the NC group (small arteries: p<0.05 for both NLFS and COPD-CS; medium arteries: p<0.05 for NLFS, p<0.01 for COPD-CS). The COPD-CS and COPD-ES groups had thicker arterial walls in large arteries than the NC group (p<0.001 and p<0.01, respectively). The COPD-CS group had the thickest arterial wall among groups across all arterial sizes, with a statistically significant difference compared with the NLFS and SAD groups in larger arteries (p<0.001 and p<0.01, respectively). There was a trend for thinner arterial walls in the COPD-ES group than in the COPD-CS group across all arterial sizes (figure 4a). Thickness was measured across three arterial layers, the intima, media and adventitia, for all arterial sizes. The intimal layer showed greater thickness in all pathological groups compared with the NC group across all arterial sizes. The medial layer showed greater thickness in the pathological groups compared with the NC group in small and medium-sized arteries. In contrast, the media of large arteries was thicker in the NLFS and COPD-CS groups than in the NC group (figure 5a–c).

a) Total arterial wall thickness in the arterial range of 100–300 µm, 300–500 µm and 500–1000 µm. b) Correlation of total arterial thickness with smoking history in current smokers with chronic obstructive pulmonary disease (COPD-CS). c) Correlation of total arterial thickness with forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) % in COPD-CS group. NC: never-smoker normal controls; NLFS: normal lung function smokers; SAD: patients with small airway disease; COPD-ES: ex-smokers with chronic obstructive pulmonary disease. *: p≤0.05; **: p≤0.01; ***: p≤0.001.

Comparison of individual layers of arterial wall between the groups for a) small arteries, b) medium arteries and c) large arteries. NC: never-smoker normal controls; NLFS: normal lung function smokers; SAD: patients with small airway disease; COPD-CS: current smokers with chronic obstructive pulmonary disease; COPD-ES: ex-smokers with chronic obstructive pulmonary disease. *: p≤0.05; **: p≤0.01; ***: p≤0.001.
Arterial remodelling, impact on lung physiology and relationship to smoking history
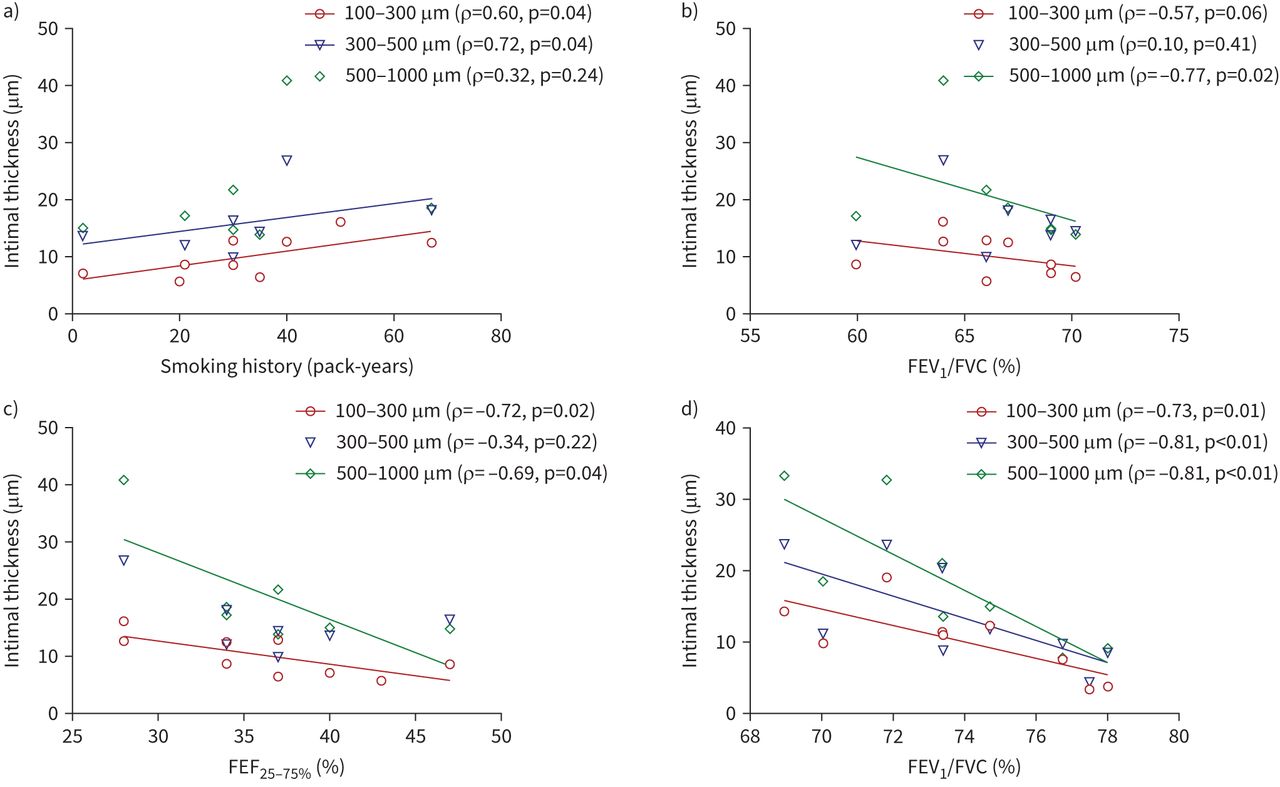
The smoking history in the COPD-CS group correlated with the greater wall thickness of medium-sized arteries (r=0.67, p=0.04) (figure 4b), while the increased wall thickness of small arteries in this group was inversely related to FEV1/FVC % (r= −0.81, p<0.01) (figure 4c). Further, the smoking history in this group was positively correlated with intimal thickness of small arteries (r=0.60, p=0.04) and medium arteries (r=0.72, p=0.04) (figure 6a). The intimal thickness in small and large arteries of this group was negatively correlated with FEV1/FVC % (small arteries: r= −0.57, p=0.06; large arteries: r= −0.77, p=0.02) and FEF25–75% (small arteries: r= −0.72, p=0.02; large arteries: r= −0.69, p=0.04) (figure 6b, c). Intimal thickness across all arterial sizes in patients with SAD was also negatively correlated with FEV1/FVC % (figure 6d).

Correlation of intima thickness in current smokers with chronic obstructive pulmonary disease (COPD-CS) with a) smoking history in pack-years, b) forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) % and c) forced expiratory flow at 25–75% of FVC (FEF25–75%). d) Correlation of intima thickness in patients with small airway disease (SAD) with FEV1/FVC %.
Elastin deposition in the pulmonary arterial wall
The inner and outer elastin layers surrounding the medial smooth muscle layers were more defined in the NC group, whereas the elastin layers were not intact in the smoking groups. Furthermore, the deposition of elastin was more pronounced within the intimal layer than in media and adventitia layers. Deposition of elastin in the small arteries of the COPD-CS group was greater than in the NC, NLFS and SAD groups (p<0.05). In contrast, deposition of elastin in the small arteries of the NLFS and SAD groups was similar to that in the NC group (figure 7a).

Comparison of sum elastin percentage for a) total arterial wall in different arterial ranges, b) individual layers in small arteries (100–300 µm), c) individual layers in medium arteries (300–500 µm) and d) individual layers in large arteries (500–1000 µm). NC: never-smoker normal control; NLFS: normal lung function smokers; SAD: patients with small airway disease; COPD-CS: current smokers with chronic obstructive pulmonary disease; COPD-ES: ex-smokers with chronic obstructive pulmonary disease. *: p≤0.05; **: p≤0.01.
For individual layers, intimal elastin deposition was greater in small arteries of the COPD-CS group than in the NC, NLFS and SAD groups (p<0.05, p<0.05 and p<0.01, respectively). In medium-sized arteries, the intimal elastin deposition in the COPD-CS group was greater than in the NLFS, SAD and COPD-ES groups (all p<0.05). In comparison, only the small arteries of the COPD-CS group showed greater media elastin deposition than in the SAD group (p=0.04). The adventitial layer of small and medium-sized arteries had greater elastin in the COPD-CS group than in the NC group (both p<0.01) (figure 7b–d).
Correlation of elastin deposition with arterial wall thickness
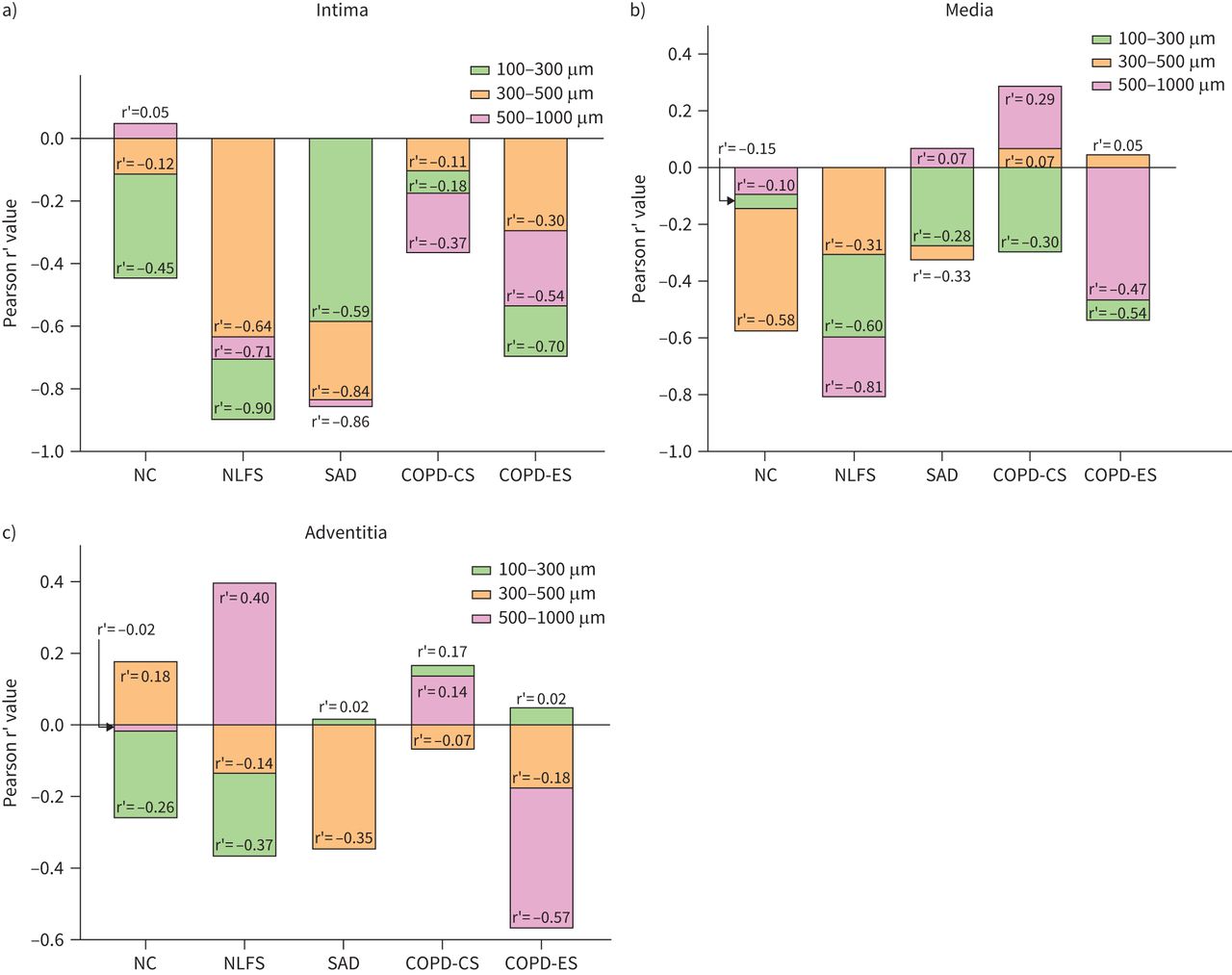
Intimal elastin deposition was negatively correlated with intima thickness for all the groups across arterial sizes, except for large arteries in the NC group. The correlation coefficient was more negative for the NLFS and SAD groups than for the COPD-CS group (figure 8a). Elastin deposition in the media of small, medium and large arteries was negatively correlated with the thickness of the media in all the study groups except for medium and large arteries in the COPD-CS group and for large arteries in the SAD group. The correlation coefficient for the media elastin and its thickness in small, medium and large arteries was more negative for the NLFS group than for the COPD-CS group (figure 8b). We observed no correlation between elastin deposition in the adventitia and thickness of the adventitia in the small, medium and large arteries of any of the study groups (figure 8c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation of thickness to elastin deposition in the a) intima, b) media and c) adventitia. Correlation calculated using Pearson's r’. The elastin deposition in the intima layer negatively correlated with thickness for all groups across sizes, except for large arteries in the never-smoker normal control (NC) group. The media layer also showed a similar trend except for large arteries in patients with small airway disease (SAD) and current smokers with chronic obstructive pulmonary disease (COPD-CS) and medium arteries in COPD-CS. No statistically significant correlations were observed for the adventitial layer. NLFS: normal lung function smokers; COPD-ES: ex-smokers with chronic obstructive pulmonary disease.
Discussion
We observed extensive vascular remodelling in both COPD and non-COPD smokers. We recorded reduced arterial number, increased arterial wall thickness and changes in elastin deposition across all arterial sizes with increased parenchymal tissue density. The COPD-CS group had the thickest arterial wall, while there was a trend for smaller thickness in the COPD-ES group compared with the COPD-CS group. An increase in intimal thickness was negatively correlated with lung function, especially in the COPD-CS and SAD groups. The results from this study also highlight the combined effect of smoking and COPD on pulmonary vascular remodelling and its contribution to changing the composition and content of the extracellular matrix environment [22].
We observed a more distorted, irregular, thickened alveolar interstitium across all pathological groups. In addition, we observed five-fold greater parenchymal tissue density in smoking groups compared with the healthy NC group, which suggests abnormal thickening of the alveolar septum. This apparent paradox of parenchymal tissue density could be explained by disproportionate thickening of the parenchymal area as an early step in smokers and patients with mild-to-moderate COPD before the actual loss of parenchymal tissue begins in severe disease stages. Results from a population-based cohort study support our findings, showing an association between the volume of high attenuation areas in computed tomography imaging and increasing smoking history, indicating subclinical parenchymal lung disease in those smokers [23]. Interestingly, in pathological groups, arterial number per parenchymal tissue density was smaller than in the non-smoking NC group. Such reduction in arterial numbers with thickened parenchymal tissue can severely affect the ventilation–perfusion ratio of the lungs [24]. Imbalance in this process in patients with mild COPD has been attributed to the disproportionately higher perfusion heterogeneity [25]. Our study also demonstrated similar parenchymal remodelling in the NLFS group, further illustrating that smoking alone can be critical to vascular modifications.
The arterial wall was thicker and the lumen was smaller across all arterial size ranges in the smoking groups, with the NLFS and COPD-CS groups having comparable thickness. Previous studies by Wrobel et al. [26] described similar increases in wall thickness of small arteries in COPD patients, while Wright et al. [27–29] reported pronounced differences in medium-large arteries of COPD patients. More mechanistic work is needed to fully understand our observation of greater changes in larger arteries of COPD patients. Larger pulmonary arteries receive the highest blood flow per cross-section area. Accordingly, we hypothesize that a reduction in the number of arteries as shown by ours and other imaging studies in COPD patients will increase blood flow per cross-sectional area in the remaining arteries. We reason that this increase in flow will increase intraluminal pressure and promote remodelling [30]. Our examination showed similar pathology in the arterial wall of smokers without COPD as with COPD patients. Further, we have shown a trend of smaller arterial wall thickness in ex- versus current smokers with COPD. The beneficial effects of smoking cessation on overall arterial stiffness are corroborated by Doonan et al. [31].
The analysis of individual arterial layers revealed a greater thickening of intima than media and adventitia across all arterial sizes in the NLFS, SAD and COPD groups than in the NC group. Previous morphometric studies showed similar trends [14, 26, 28], e.g. Wright et al. [28, 29] reported greater intima thickness of smokers with moderate and severe airway obstruction and in media with severe obstruction. Similar findings by Santos et al. [14] support our result on thickened intima in COPD and non-COPD smokers. However, quantification of the arterial remodelling changes for this range of arterial sizes in five different patient cohorts is unique to our study. Hypoxia has been classically considered as the major pathogenic mechanism in arterial remodelling [17]. Hypoxia-inducible factor 1α has been reported to increase in patients with COPD [32]. However, our observations of a similar degree of remodelling in COPD and non-COPD smokers across arterial sizes suggest predominantly cigarette smoke-induced endothelial dysfunction. These cigarette smoke-induced changes may explain the arterial remodelling observed in smokers and patients with mild-to-moderate COPD, although it might be insufficient to produce PH, which mainly occurs in advanced COPD stages but we believe has an early onset [13, 33]. Therefore, we speculate that hypoxia contributes to the endothelial dysfunction in advanced COPD. The prominent finding of the proliferation of intima with aggressive encroachment into luminal space in our study can be attributed to the ability of the endothelial cell to transform into a more proliferative phenotype, i.e. endothelial-to-mesenchymal transformation; however, its extent and molecular mechanism need further exploration [34–36].
Another novel contribution of our study is the inclusion of patients with SAD [20, 37, 38]. In these patients, we recorded a positive association between airway obstruction and arterial remodelling. This finding suggests early onset of arterial remodelling that is reminiscent of early-onset smoke-associated insult to the small airways. A previous human study also found that vascular changes correlate highly with small airway pathology [39], though it was limited by both group and size comparisons. In vivo studies have supported the possible link between small airway inflammation and vascular changes [28, 40].
We further investigated the contribution of extracellular matrix protein elastin in arterial layers. The structural integrity and elasticity of normal pulmonary arteries are maintained by the elastin fibres, which are arranged as internal and external laminas. Therefore, disruption in the elastin fibres has significant consequences on arterial remodelling and the development of PH [41]. Our study showed considerable elastin deposition in the COPD-CS group, most evident in the intimal layer of smaller and medium-sized arteries. Our findings are similar to those of Ferrer et al. [42], who demonstrated the role of hypoxia in altering elastin deposition. We also observed that elastin deposition was greater in the COPD-CS patients than in the NLFS and SAD groups. The disparity of elastin deposition in our study could be explained by the initial destruction of elastin due to cigarette smoke exposure in the NLFS group, which returns to normal in COPD smokers with stimulated connective tissue synthesis [43]. However, the resynthesised elastic fibres are disorganised and defective, and the normal elastic network cannot be restored because of their size and molecular complexity [44]. Santos et al. [14] reported the lack of correlation between intimal enlargement and intimal elastin deposition in patients with mild COPD. In addition, our correlation coefficient findings for thickness versus elastin deposition were more negative for the NLFS and SAD groups than for the COPD group. These differences in elastin deposition in COPD and non-COPD suggest separate mechanisms contributing to elastin turnover.
Smoking history affected arterial wall thickness, particularly the intima in the COPD-CS group. In addition, arterial wall thickness of smaller arteries was negatively correlated with FEV1/FVC % and FEF25–75%. The negative correlation between the wall thickness of the small arteries and the severity of obstructive lung disease suggests that small arteries and small airways are the initial and most affected sites of inflammation, with possible perivascular and interstitial involvement [28, 45]. Wilkinson et al. [46] and Wright and Churg [28] found a robust negative correlation of pulmonary arterial pressure and with FEV1 and FEF25–75%. Similarly, Hale et al. [39] showed that the vascular changes were highly correlated with emphysema and small airway pathology. Our findings, in line with previous studies, suggest a concomitant effect of smoking on pulmonary vasculature and airways.
The limitations of this study include the small sample size for each group; however, the data lacked significant intragroup variability, and our findings were statistically robust throughout. In addition, we had no computed tomography scan images. This is a limitation for explaining the parenchymal thickening; yet, our histopathology data indicate that parenchymal thickening might have been due to a mild-to-moderate form of COPD and damage may be early in onset. Subjects in the NC group were about 20 years younger than subjects in other groups; however, no association between arterial remodelling and age could be established in these subjects. In addition, all our pathological samples were from lung cancer patients, and we may not be able to rule out this effect completely; however, all our tissue sampling was done well away from cancer-affected areas. Also, due to unavailability of cardiac function data in patients with COPD, we could not establish an association between arterial remodelling and PH in this study.
In summary, this study provides morphometric data on the remodelling of muscular arteries in different cohorts of smokers and COPD patients. More importantly, we found that smoking, SAD and mild-to-moderate COPD were associated with pruning of and a decrease in the number of pulmonary vessels, with increased wall thickness and with variable elastin deposition. These changes were associated with worse airway obstruction and could be critical to the early development of PH. We recorded similar vascular changes in subjects with a passive smoking history as well, suggesting ill effects of passive smoking at a pathological level. Further, our results also suggest the positive outcome of smoking cessation with lesser remodelling changes in ex-smokers. Our data support the need to detect early vascular changes in smokers and COPD patients and to design appropriate therapeutic options to modify the disease trajectory, which would otherwise likely progress to PH and its complications.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: S.S. Sohal reports personal fees for lectures from Chiesi outside the submitted work. The other authors do not have any conflict of interest to declare.
Support statement: This work was supported by research grants from Clifford Craig Foundation, Launceston General Hospital, Rebecca L. Cooper Medical Research Foundation, Royal Hobart Hospital Research Foundation, Cancer Council Tasmania and TSANZ Boehringer Ingelheim COPD Research Award. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received May 23, 2022.
- Accepted August 15, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










