





Abstract
Purpose Obstructive lung disease is increasingly common among persons with HIV, both smokers and nonsmokers. We used aptamer proteomics to identify proteins and associated pathways in HIV-associated obstructive lung disease.
Methods Bronchoalveolar lavage fluid (BALF) samples from 26 persons living with HIV with obstructive lung disease were matched to persons living with HIV without obstructive lung disease based on age, smoking status and antiretroviral treatment. 6414 proteins were measured using SomaScan® aptamer-based assay. We used sparse distance-weighted discrimination (sDWD) to test for a difference in protein expression and permutation tests to identify univariate associations between proteins and forced expiratory volume in 1 s % predicted (FEV1 % pred). Significant proteins were entered into a pathway over-representation analysis. We also constructed protein-driven endotypes using K-means clustering and performed over-representation analysis on the proteins that were significantly different between clusters. We compared protein-associated clusters to those obtained from BALF and plasma metabolomics data on the same patient cohort.
Results After filtering, we retained 3872 proteins for further analysis. Based on sDWD, protein expression was able to separate cases and controls. We found 575 proteins that were significantly correlated with FEV1 % pred after multiple comparisons adjustment. We identified two protein-driven endotypes, one of which was associated with poor lung function, and found that insulin and apoptosis pathways were differentially represented. We found similar clusters driven by metabolomics in BALF but not plasma.
Conclusion Protein expression differs in persons living with HIV with and without obstructive lung disease. We were not able to identify specific pathways differentially expressed among patients based on FEV1 % pred; however, we identified a unique protein endotype associated with insulin and apoptotic pathways.
Abstract
BALF protein profile distinguishes HIV-associated obstructive lung disease. A unique endotype in black men with more severe obstructive lung disease was found that associated with signal transduction pathways, cell cycle and apoptosis. https://bit.ly/3u7j3Np
Introduction
Improved survival in persons with HIV has led to a higher prevalence of several chronic illnesses including obstructive lung disease, which affects an estimated 3–23% [1–8]. A major gap in knowledge is the inability to identify those at risk and a limited understanding of why HIV increases obstructive lung disease risk independent of smoking status. In the era prior to the common use of combination antiretroviral therapy pulmonary obstruction was mostly associated with advanced HIV/AIDS and frequent pulmonary infections [9]. In the antiretroviral treatment era, obstructive lung disease persists as a frequent comorbidity even in the absence of AIDS or frequent pulmonary infections [1–5, 8, 10–14]. While pulmonary infections may still have some role in obstructive lung disease development in persons living with HIV [15], it is highly likely that other factors are involved in HIV-associated obstructive lung disease, including the HIV virus itself. This is particularly pertinent as the lung is a reservoir for HIV and site for HIV replication [16]. Currently no biomarker identifies risk or lends insight into the mechanisms that lead to a rapid decline of lung function and subsequent obstructive lung disease in persons living with HIV.
The goal of this study was to identify lung-specific biomarkers and corresponding biological pathways associated with HIV-associated obstructive lung disease using aptamer-based assays to profile the proteome in bronchoalveolar lavage fluid (BALF) in persons living with HIV comparing those with and without obstructive lung disease. Included in our analysis is targeted, mass spectrometry-based metabolite analysis. We employed statistical and computational methods to identify endotypes and proteomic pathways to lend insight into putative mechanisms of disease.
Methods
We performed a cross-sectional, matched case–control study using BALF and plasma samples previously collected from two cohorts.
Study population
Cases and controls were persons living with HIV selected from the Pittsburgh and Vancouver Lung HIV Cohorts [17, 18]. We identified 26 cases with HIV and obstructive lung disease with available BALF; obstructive lung disease was defined as the ratio of forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC) <lower limit of normal. Pulmonary function tests were obtained within 3 months of acquiring the BALF. Participants fasted prior to BALF collection, while the majority, but not all, fasted prior to plasma collection. Sample size was based on our previously reported study [19]. Controls consisted of 26 individuals with HIV and normal lung function (defined as FEV1/FVC >lower limit of normal and FEV1 >80% of predicted normal) matched on age (±5 years), antiretroviral treatment use and smoking status (current versus nonsmoker). Participants in the parent cohort studies provided informed consent for BALF collection and storage; the current study was approved by the University of Minnesota Institutional Review Board. Parent studies had Institutional Review Board Approval from Pittsburgh and Vancouver.
Sample processing
At study enrolment, BALF was collected as previously described [17, 18]. Samples were stored at −80°C prior to processing and underwent one freeze–thaw cycle. BALF samples were vortexed and centrifuged at 5000 ×g for 5 min at 4°C followed by separation of the pellet and supernatant for the removal of additional debris. Samples were prepared for proteomics following SomaScan® Assay specification, specifically 75 μL of sample concentrated to 200 μL·mL−1. To identify metabolites, 10 μL of plasma or 200 μL of BALF were loaded onto a Biocrates Life Sciences Absolute IDQ p400 HR (Biocrates Life Sciences catalog number 21018; Biocrates Life Sciences, Innsbruck, Austria) following the manufacturer's instructions and as previously reported [20]. Metabolite analysis was performed on a Thermo Scientific Q Exactive TM, Hybrid Quadrupole-Orbitrap TM (Thermo Fisher Scientific, Waltham, MA, USA), mass spectrometer equipped with a Thermo Scientific Ultimate 3000 UHPLC equipped with an autosampler, following manufacturer parameters.
Data cleaning
The SomaScan® proteomics assay data contained 7335 aptamers targeting human proteins. For each aptamer, we calculated an empirical lower limit of detection (LOD) to filter out those with over 50% of samples below the LOD. We calculated the LOD as  where
where  is the median absolute deviation between each sample and the median aptamer level [21]. LODs were calculated using the detected aptamer levels from buffer samples. We removed 3048 aptamers with over 50% of samples below the corresponding empirical LOD, retaining 4253 aptamers. We used Fisher's exact test to assess if the aptamers removed during this procedure are over-represented in cases versus controls and found no significant difference in samples above or below the LOD between cases and controls after adjusting for multiple comparisons. The remaining aptamers mapped to 3872 unique protein targets by the UniProt ID [22]. For the BALF metabolomic data we removed any metabolites that were either missing data or below the LOD for >50% of the cohort leaving 252 metabolites. There were 258 metabolites for plasma. We applied a log(1+x) transformation to both datasets, scaled and centred each protein and metabolite to have mean 0 and standard deviation 1.
is the median absolute deviation between each sample and the median aptamer level [21]. LODs were calculated using the detected aptamer levels from buffer samples. We removed 3048 aptamers with over 50% of samples below the corresponding empirical LOD, retaining 4253 aptamers. We used Fisher's exact test to assess if the aptamers removed during this procedure are over-represented in cases versus controls and found no significant difference in samples above or below the LOD between cases and controls after adjusting for multiple comparisons. The remaining aptamers mapped to 3872 unique protein targets by the UniProt ID [22]. For the BALF metabolomic data we removed any metabolites that were either missing data or below the LOD for >50% of the cohort leaving 252 metabolites. There were 258 metabolites for plasma. We applied a log(1+x) transformation to both datasets, scaled and centred each protein and metabolite to have mean 0 and standard deviation 1.
Statistical analysis
We assessed the ability of the proteomic results to separate cases from controls using sparse distance-weighted discrimination (sDWD) [23], a method to classify subjects based on a large number of features (i.e., proteins) in the setting when the data are high-dimensional, or when the number of features exceeds the number of samples. Cases were labelled as 1 while controls were labelled as −1. sDWD calculates a score, a linear combination of protein expression levels, for each observation that captures how confidently cases and controls can be classified based on protein expression: highly positive scores for cases and highly negative scores for controls reflects good separation between the two classes. An L1 penalty on the coefficients of the proteins induces variable selection. We used cross-validation where we iteratively held out each case–control pair as a test set and trained the sDWD model on the remaining pairs. We compared the average, cross-validated sDWD scores for cases and controls using a paired t-test and the area under the curve (AUC) to assess the overall performance of the classifier. We used the sdwd R package version 1.0.5 by the authors [23] to implement this method.
We used the Global Lung Initiative standards to calculate FEV1 per cent predicted (FEV1 % pred) [24]. To investigate the relationship between each individual protein and FEV1 % pred we used the Pearson correlation in a permutation testing framework to ensure our results were robust to deviations from normality, and to accommodate scenarios in which multiple aptamers map to the same protein. We first used a correlation test for each aptamer and FEV1 % pred and combined the p-values for aptamers that mapped to the same protein using Fisher's method [25]. This yielded a chi-squared test statistic for each protein. Then, for 10 000 permutation iterations, we permuted the FEV1 % pred values across the patient cohort, applied the t-test for the correlation between each aptamer and FEV1 % pred under the permuted labelling scheme, and combined resulting p-values for aptamers mapping to the same protein using Fisher's method to obtain a chi-squared test statistic for each protein. This resulted in 10 000 combined chi-squared test statistics for each unique protein target. We computed a permutation p-value for each protein by taking the proportion of chi-squared test statistics that exceeded the chi-squared test statistic based on the original FEV1 % pred observations. We applied a false discovery rate (FDR) correction [26] to the permutation p-values to correct for multiple comparisons. The proteins that were significant at the 0.05 level after permutation testing prior to multiple comparisons adjustment were then used in a pathway over-representation analysis using IMPaLA software [27]. We considered an analogous permutation testing framework to study the correlation between each protein and FEV1/FVC and diffusing capacity of the lung for carbon monoxide (DLCO) % predicted.
We identified protein-driven endotypes using K-means clustering. We ran the K-means algorithm 100 times with 10 random start points. For each of the 100 K-means replications, we saved the clustering scheme that achieved the minimum within-cluster variability across the 10 random start points to ensure an optimal solution [28]. We tested for individual protein differences between the clusters using the permutation testing framework described previously. At each permutation iteration, we permuted the cluster labels across the patient cohort and compared the permuted clusters using a t-test. Proteins that were significantly different between the clusters at an FDR level of 0.05 were used in pathway over-representation analysis. To quantify our uncertainty of the K-means clusters, we considered K-means clustering with 100 bootstrapping replications using the bootcluster package [29] in R.
For comparison, we also applied the same K-means clustering approach on metabolomics data collected from BALF and plasma in the same patient cohort. We tested for individual metabolite differences between the resulting clusters and quantified our uncertainty surrounding these clusters using bootstrapping.
Results
Study participant characteristics
Table 1 summarises the demographics of the individuals in our study. This cohort consisted largely of males (73.1%) with a mean age of 56.7 years. Over half (53.8%) of individuals identified as black and non-Hispanic and the same percentage identified as a current smoker (mean pack-years 23.1). Lung function (FEV1) ranged from 21 to 90% of predicted normal in the obstructive lung disease cases (all of whom had FEV1/FVC <lower limit of normal), compared to from 80 to 128% in the controls. DLCO did not differ amongst the ∼80% of cases and controls that had it measured. Most individuals (92.3%) were treated with antiretroviral treatment at the time of sample collection. Two individuals among the cases and three among the controls exhibited viral loads >50 copies·mL−1, among those for whom we had available viral load data.
Demographics of the patient cohort considered in this study
BALF proteome differences in obstructive lung disease
We used sDWD to assess the collective power of BALF proteins to distinguish between cases and controls. This analysis shows a significant difference in the measured proteome between participants with obstructive lung disease and those without. Figure 1 displays the distribution of sDWD scores for cases and controls with cross-validation. Large differences in the average sDWD scores for cases and controls suggests better prediction accuracy based on protein expression. We compared the average scores for cases and controls under cross-validation using a paired t-test, which yielded a p-value of 0.0027 and an AUC of 0.6538.
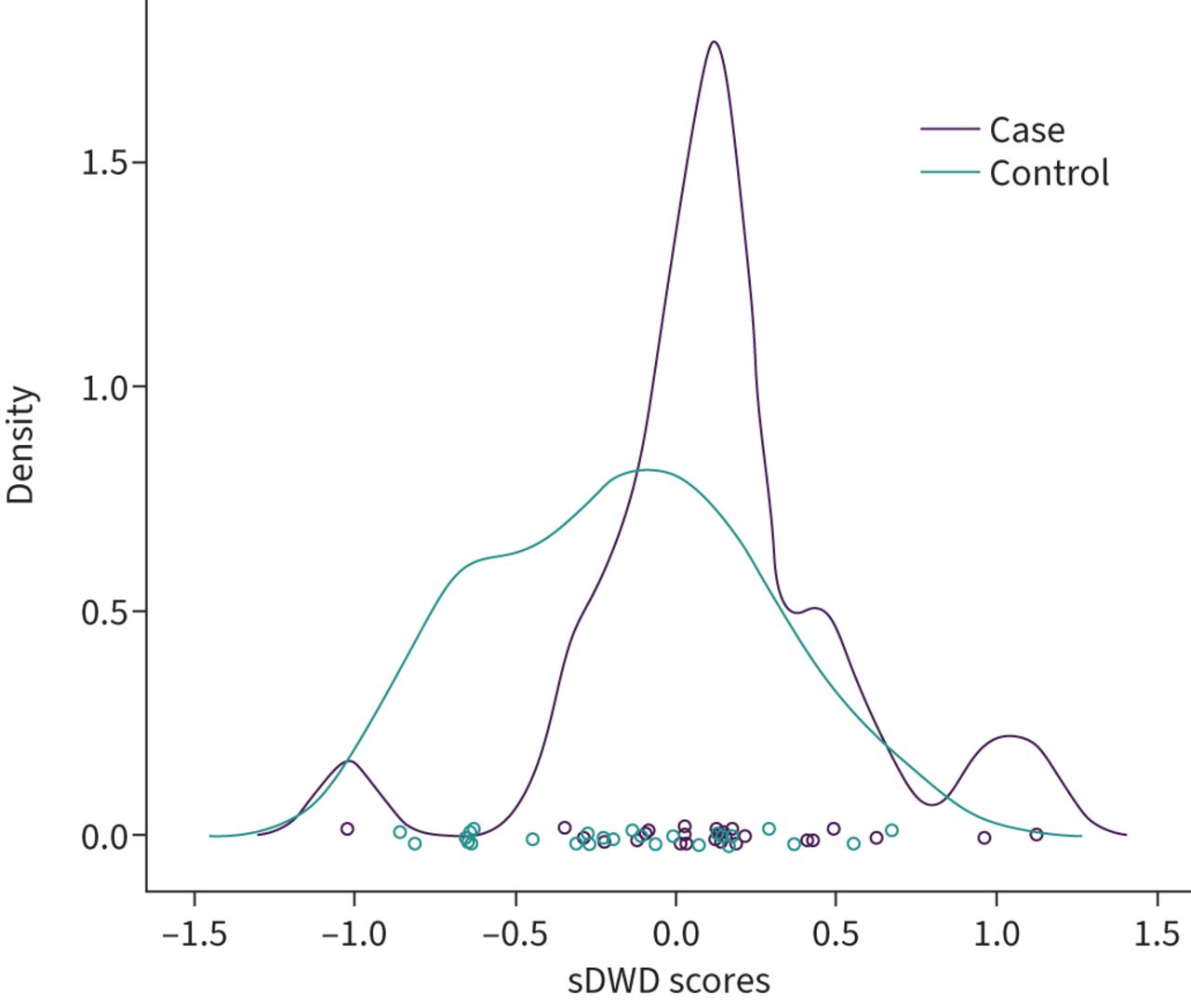
Densities of sparse distance-weighted discrimination (sDWD) scores for cases and controls based on bronchoalveolar lavage fluid protein expression. sDWD scores are a linear combination of protein expression levels. Distinct separation in the scores between cases and controls reflects more power to distinguish the classes based on protein expression.
We then considered the correlation between FEV1 % pred and each BALF protein within a permutation testing framework. Table 2 summarises the top proteins most significantly correlated with FEV1 % pred. We found that 1305 proteins were significantly correlated with FEV1 % pred at the 0.05 level, prior to FDR adjustment and 575 proteins were significant at the 0.05 level after FDR adjustment. Proteins significant at the 0.05 level, prior to adjustment, were filtered into pathway over-representation analysis using IMPaLA software. Although we found many significant proteins, no pathways met significance after controlling for multiple comparisons (supplementary table 1S).
Top 11 proteins most significantly correlated with FEV1 % pred
We found 1467 proteins were significantly correlated with FEV1/FVC at the 0.05 FDR level (supplementary table 2S). No proteins were significantly correlated with DLCO % pred after multiple comparisons adjustment.
Endotypes identified by cluster analysis
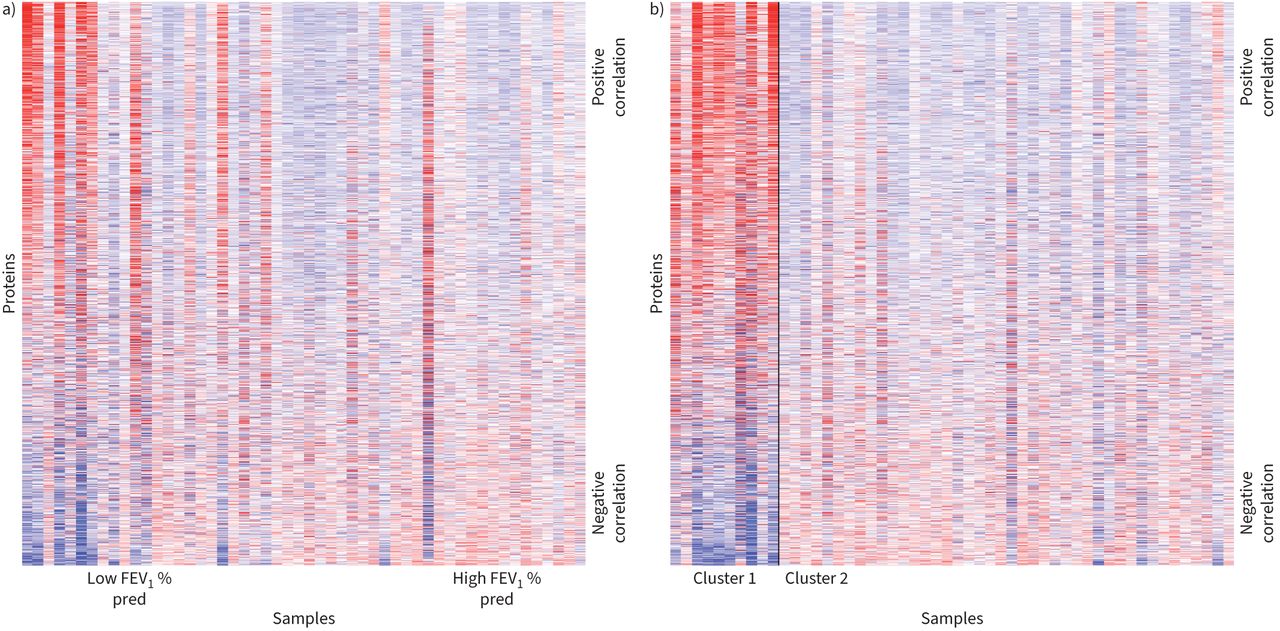
Heatmaps of the protein expression revealed a visually distinct subgroup of patients who tended to have lower FEV1 % pred values (figure 2a). We thus constructed protein-driven endotypes using K-means clustering with 100 replications for K=2 clusters. K-means clustering results were stable across the 100 replications, with each replication yielding the same clustering scheme of 10 individuals in one cluster and 42 in the other, referred to as Cluster 1 and Cluster 2, respectively. Figure 2b shows the protein expression across the patient cohort, with observations grouped by their assigned cluster. Table 3 demonstrates that Cluster 1 was largely male (80%) and on average older than Cluster 2 (62.5 versus 55.3 years). Cluster 1 also largely comprised individuals who identified as black (70%) compared to Cluster 2 where 50% of individuals identified as black. Cluster 1 also comprised individuals with lower average FEV1 % pred (63.9 versus 91.0) and lower average FEV1/FVC (0.514 versus 0.714), and 90% of individuals were diagnosed with obstructive lung disease compared to 40.5% in Cluster 2. DLCO was available for six out of 10 individuals in Cluster 1 and 35 out of 42 in Cluster 2. The average DLCO in Cluster 1 was 0.611 and 0.764 in Cluster 2 (p=0.15). The overall stability of this clustering scheme, as determined under bootstrap replications, was 89%.

Heatmaps show protein expression across individuals in study cohort. Columns reflect samples while rows reflect proteins. In both heatmaps, proteins are ordered based on the direction and magnitude of their correlation with forced expiratory volume in 1 s % predicted (FEV1 % pred). a) Heatmap samples ordered by FEV1 % pred. b) Samples are grouped within their respective K-means clusters and the solid black line separates the two groups. Within clusters, samples are ordered by FEV1 % pred.
Demographics of two K-means clusters determined using protein expression
We used an analogous permutation testing scheme to compare proteins across the clusters. The top 10 proteins that were significant at an FDR level of 0.05 are shown in table 4, and all 1279 significant proteins after FDR adjustment were used for pathway analysis. The top pathway distinguishing these two clusters involved insulin, with an FDR-adjusted p-value of 0.0312 (table 5). Other top pathways involved FOXO transcription factors, apoptosis, RNA metabolism and retinol metabolism.
Top 10 proteins significantly different between two K-means clusters determined based on protein expression
Top nine pathways based on proteins that were differentially expressed between K-means clusters and were significant at an FDR level of 0.05
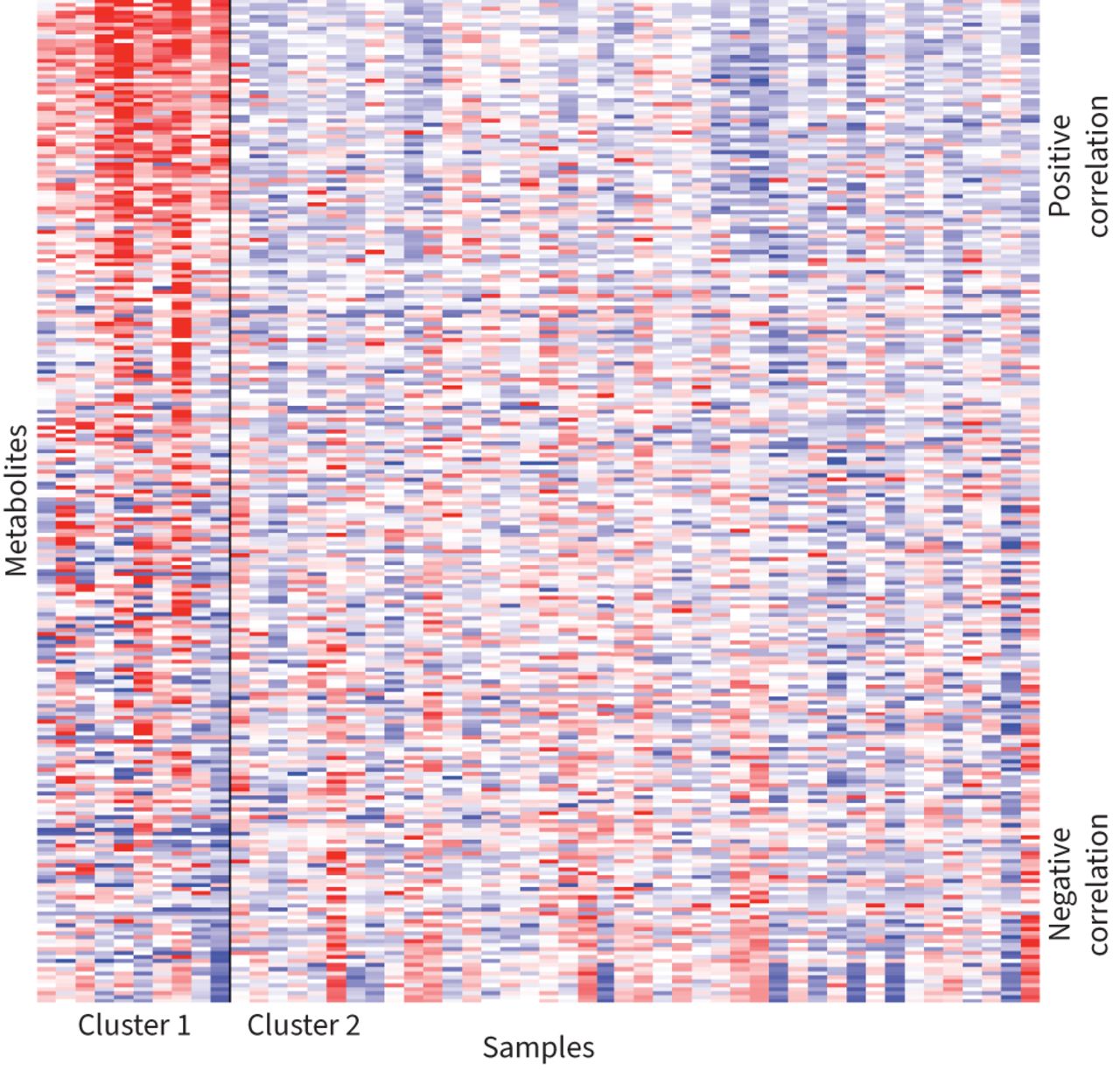
To determine if the proteomic findings extended to metabolomic expression we performed a K-means clustering on the BALF metabolomics data. We found that the metabolomic expression yielded similar results to the BALF proteomics data with similar heatmaps of metabolite expression (figure 3). We obtained a consistent clustering scheme across 100 replications: one cluster with 10 individuals and another cluster with 42 (supplementary table 4S). However, no cluster was identified using plasma metabolites (supplementary figure 1S). Asparagine was the most significant BALF metabolite in cluster 1, while overall acylcarnitines were the predominant metabolites in this cluster (supplementary table 5S). While the cluster sizes were identical to those obtained using the BALF protein expression data, the composition of the metabolite clusters differed slightly. There was an overlap of six individuals in the smaller cluster of 10 between the protein-driven and metabolite-driven clusters (supplementary table 6S). These six individuals were all male with an average age of 65.7 years and all identified as black.

{kind=link}
{kind=link}
{kind=link}
Heatmap shows metabolite expression for the study cohort. Columns reflect samples and rows reflect metabolites. The metabolites are ordered by the direction and magnitude of their correlation with forced expiratory volume in 1 s % predicted. The samples are grouped within their respective K-means clusters and the solid black line separates the two groups.
Discussion
We found that the BALF proteome in persons living with HIV distinguishes those with obstructive lung disease from those without obstructive lung disease. However, we did not identify any pathways composed of proteins that were differentially expressed among individuals with high FEV1 % pred and those with low FEV1 % pred. We did identify an endotype driven by both proteomic and metabolomic BALF molecular contributors, but not plasma metabolites. Based on the differentially expressed BALF proteins, this endotype exhibited over-representation of insulin- and apoptosis-related pathways, suggesting that signal transduction pathways with FOXO and cell cycle are important regulators.
Previous studies identified plasma proteins associated with pulmonary dysfunction in HIV. These include elevated plasma interleukin (IL)-6, C-reactive protein, endothelin-1 [30] and activation of inflammatory pathways [31, 32]. We found similar findings in the START cohort where higher plasma levels of IL-6, high-sensitivity C-reactive protein and serum amyloid A associated with lower FEV1 and FVC [33]. In addition, unique phenotypes associated with inflammatory pathways have been identified by cluster analysis in HIV-associated obstructive lung disease [34]. Unlike our current study, these studies were obtained from blood samples, not direct sampling of the lung. Since the BALF proteome in healthy persons living with HIV differs significantly from non-HIV controls [35], we sought to identify lung-specific biomarkers of HIV-associated obstructive lung disease.
This study was designed as a case–control study (obstructive lung disease present/absent) where pairs were matched based on age, antiretroviral treatment status and smoking status. Though we found the BALF proteome moderately differentiates cases and controls, we primarily considered FEV1 % pred as an outcome rather than case–control status due to the heterogeneity in lung function within each group, which also improved study power. We found 1305 proteins that were significantly correlated with FEV1 % pred at the 0.05 level and 575 proteins that were significant after FDR adjustment. Our finding that BALF IL-18 Ra is highly correlated with FEV1 % pred is consistent with Imaoka and colleagues [36] who reported IL-18 as highly expressed in lung tissue among COPD patients without HIV and that increased expression associated with a decrease in FEV1 % pred. This has not been previously reported in obstructive lung disease associated with HIV. We also found the protein ephrin-A2 to be highly associated with FEV1 % pred and its gene, EFNA2, is associated with weight loss among patients with non-HIV COPD [37]. Despite identifying many proteins significantly correlated with FEV1 % pred, we were not able to detect pathways reflected by these proteins. This may be due to lack of statistical power in our relatively small sample size.
We identified two endotypes driven by proteomic expression in BALF that were also identified in the BALF metabolome. These BALF endotypes were consistently detected across 100 replications with 10 random start values of the K-means clustering algorithm, suggesting these clusters are robust to many different initialisations of the algorithm. We detected these endotypes in both the BALF proteome and the metabolome but not the plasma metabolome, though the compositions of the clusters differed slightly between the BALF proteomic and metabolomic platforms. Both the metabolome and the proteome had a smaller endotype consisting of 10 individuals, which showed clear differential expression in heatmaps. There were six individuals who were consistently grouped into this smaller cluster between both sources, suggesting they possess a unique BALF expression profile apparent in both the proteome and metabolome (supplementary table 6S). These six individuals all identified as black non-Hispanic males who met the criteria for obstructive lung disease and exhibited lower FEV1 % pred compared to Cluster 2.
Pathway analysis using the significant proteins identified in cluster 1 revealed several pathways. Among those were insulin, regulation of FOXO transcription factors and apoptosis pathways. Many of the proteins expressed in these pathways are involved in signal transduction and cell cycle regulation. Two promoter forkhead box (FOX) proteins were among the proteins in the insulin and FOXO pathways. FOX promoter expression is increased in epithelial cells in non-HIV COPD patients; however, in patients with mucus hyperexcretion phenotype it is depleted. This transcription factor is involved in goblet cell differentiation in the airway epithelium, and aberrant methylation patterns have been described in non-HIV COPD lung epithelium [38–41].
Pathway analysis of our endotype also revealed an intrinsic pathway for apoptosis as being significant. Enhanced apoptosis in lung endothelial and epithelial cells is found in non-HIV COPD and is felt to be a critical step in COPD pathogenesis [42, 43]. In non-HIV COPD, enhanced apoptosis has been associated with the emphysema phenotype. Computed tomography imaging was not available to quantify emphysema in our study and the DLCO, which correlates with the emphysema phenotype, did not vary between endotypes, although the relatively small sample size of Cluster 1 likely limited statistical power. Accelerated apoptosis is one mechanism proposed for the loss of CD4+ T-lymphocytes in HIV infection [44]. It is also postulated that HIV-infected persons have increased susceptibility to apoptosis because the HIV proteins Tat and Nef induce endothelial cell apoptosis [45, 46]. Further investigations are necessary to determine whether these apoptotic pathways are associated with lymphocyte or lung parenchymal cells.
Our study has a few limitations. Owing to our small sample size and large number of proteins, we were unable to detect any significantly over-represented pathways reflected by proteins associated with FEV1 % pred. In a future study with more participants, we may have more power to identify pathways associated with lung function decline. It would be interesting to recruit HIV-negative controls to assess if differentially expressed proteins and protein-driven pathways are unique to HIV-modulated obstructive lung disease or are exhibited across the population of patients with obstructive lung disease. A larger sample size and validation cohort would also be beneficial to corroborate the endotypes we detected in our study and their clinical outcomes. Though our study was a matched case–control study, individuals were not matched based on race or sex, and additional studies would be necessary to validate if these findings are race- or sex-specific. Though pairs were matched based on smoking status, we did not account for this in our correlation analysis, which is a limitation. Although not a limitation of the study, we did not find differences reflected in the plasma metabolome, thus limiting the feasibility in applying these BALF findings as global biomarkers of lung disease. In addition, a longer, longitudinal study in which protein expression is measured over time may highlight proteins that are relevant to lung function change, as previous research has shown that proteins associate differently with COPD outcomes at a single time point versus longitudinally [47].
In conclusion, the proteomic BALF profile distinguishes HIV-associated obstructive lung disease. Furthermore, a unique endotype was identified in both BALF proteomic and metabolomic profiles that predominantly were in black men with more severe obstructive lung disease, and this cluster was not found in the plasma metabolome. The proteomic pathways that were differentially expressed within this endotype were linked to signal transduction pathways, cell cycle and apoptosis.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00332-2022.SUPPLEMENT
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: No conflict of interest related to the work in this manuscript for any of the authors. K.M. Kunisaki reports consulting (Allergan/AbbVie), and independent data safety and monitoring boards (Nuvaira and Organicell), outside of this work.
Support statement: Supported by National Institutes of Health grant R01 HL140971-01A1 (all authors). This material is also the result of work supported with resources and the use of facilities at the Minneapolis Veterans Affairs Medical Center, Minneapolis, MN, USA. The views expressed in this article are those of the authors and do not reflect the views of the US Government, the Department of Veterans Affairs, the funders, the sponsors or any of the authors’ affiliated academic institutions. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received July 12, 2022.
- Accepted November 24, 2022.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










