





Abstract
Aims Oxidative stress is an important amplifying mechanism in COPD; however, it is unclear how oxidative stress changes and what its exact amplification mechanism is in the pathological process. We aimed to dynamically analyse the progression of COPD and further elucidate the characteristics of each developmental stage and unveil the underlying mechanisms.
Methods We performed a holistic analysis by integrating Gene Expression Omnibus microarray datasets related to smoking, emphysema and Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification based on the concept of gene, environment and time (GET). Gene ontology (GO), protein–protein interaction (PPI) networks and gene set enrichment analysis (GSEA) were used to explore the changing characteristics and potential mechanisms. Lentivirus was used to promote HIF3A overexpression.
Results In smokers versus nonsmokers, the GO term mainly enriched in “negative regulation of apoptotic process”. In later transitions between stages, the main enriched terms were continuous progression of “oxidation-reduction process” and “cellular response to hydrogen peroxide”. Logistic regression showed that these core differentially expressed genes (DEGs) had diagnostic accuracy in test (area under the curve (AUC)=0.828) and validation (AUC=0.750) sets. GSEA and PPI networks showed that one of the core DEGs, HIF3A, strongly interacted with the ubiquitin-mediated proteolysis pathway. Overexpression of HIF3A restored superoxide dismutase levels and alleviated the reactive oxygen species accumulation caused by cigarette smoke extract treatment.
Conclusion Oxidative stress was continuously intensified from mild emphysema to GOLD 4; thus, special attention should be paid to the identification of emphysema. Furthermore, the downregulated HIF3A may play an important role in the intensified oxidative stress in COPD.
Abstract
Oxidative stress is especially intensified from mild emphysema to GOLD 4 and downregulated HIF3A may contribute to this https://bit.ly/3FjUhQW
Introduction
COPD is a common chronic disease characterised by persistent respiratory symptoms and airflow limitation, which remains a significant global health problem due to its high prevalence and mortality [1]. A Global Burden of Disease (GBD) study reported that the disease resulted in the death of an estimated 3.2 million people worldwide in 2017 [2]. In China, the prevalence of COPD among adults aged 20 years and older is 8.6%, and the prevalence among adults aged 40 years and older is 13.7%. This indicates that nearly 100 million people have COPD in China [3]; thus, improving the prevention and treatment of this disease is critical.
In most patients with COPD, the disease develops over decades and involves aetiology, pathology and the worsening of clinical symptoms [4–6]. Several studies have shown that the late diagnosis of COPD is the most important factor underlying poor prognosis among a significant proportion of patients [7–9]. Therefore, it is not sufficient to confine the study of COPD to the stage of clinical symptoms; the focus should be shifted forward to the period of development of pathological lesions, such as emphysema (the typical pathogenic development phase of COPD), and the period involving the risk factor of smoking. A holistic and dynamic analysis of COPD, taking into account smoking, emphysema and Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification, which reflects the clinical symptoms, is needed.
Recently, a new concept has emerged, which integrates gene (G), environment (E) and clinical phenotype information from basic transcriptomics over time (T). GET was a novel approach which, by integrating information from basic and clinical omics with exposures over the lifespan, attempted to uncover novel opportunities for prevention and early treatment of COPD, leading to improved understanding of underlying pathogenic mechanisms and identification of novel targets [10]. There have been many transcriptomics studies comparing COPD patients and healthy individuals [11–13]. However, thus far, no study has combined data related to aetiology, pathology and GOLD classification to conduct a holistic and dynamic analysis of COPD. In this study we conducted a GET analysis of COPD by integrating the transcriptomics microarray data, including smoking (the aetiology of and environmental pollutants in COPD), emphysema (the clinical phenotype and progression stage of COPD) and the GOLD classification (indicating the worsening of clinical symptoms of COPD), in order to elucidate the characteristics in each of the progression stages and unveil the underlying mechanisms.
Materials and methods
Dataset preparation
Microarray datasets were screened from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo). The selection criteria were as follows: 1) lung tissue samples from nonsmokers, smokers and emphysema and COPD patients were included; 2) former smokers who had quit smoking when participating in the programme were included; 3) diffusing capacity of the lung for carbon monoxide (DLCO) <80% predicted and/or emphysema shown by computed tomography were included; 4) the classification of COPD should be according to the GOLD guidelines; and 5) patients with asthma who had a persistent airflow obstruction were excluded. Based on these criteria [1, 14–16], the GSE37768 (smokers versus nonsmokers), GSE119040 (emphysema versus non-emphysema patients), GSE1650 (severe versus mild emphysema patients) and GSE69818 (GOLD 4 versus GOLD 1 COPD patients) datasets were obtained. The sample types and platform information of each dataset are described in table 1.
Selection of GEO datasets on smokers versus nonsmokers, emphysema versus non-emphysema patients, severe versus mild emphysema patients and GOLD 4 versus GOLD 1 patients
Differentially expressed gene identification and enrichment analysis
To identify the differentially expressed genes (DEGs), GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was used with each dataset. GEO2R is an interactive online tool that compares two groups of samples collected under the same experimental conditions [17]. Adjusted p<0.05 and |log2fold change| ≥0.5 were used as cut-off values. Gene ontology (GO) analysis was used to investigate the biological functions of the DEGs, which involved determining the enriched biological process, cellular component and molecular function terms using the functional annotation tool in the DAVID website [18, 19]. The top 10 terms were visualised using ggplot2 v3.3.2.
Logistic regression prediction model based on the core DEGs
We used GSE37768 data as the training set and GSE8581 data as the test set, which included 18 lung tissue samples from control subjects (forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC)>0.7, FEV1 % pred >80%) and 15 lung tissue samples from COPD subjects (FEV1/FVC <0.7, FEV1 % pred <70%). Receiver operating characteristic curves were used to visualise the model, area under the curve (AUC) was used to evaluate its diagnostic ability, and the Youden index was used to evaluate its sensitivity and specificity.
Construction of the protein–protein interaction networks
A protein–protein interaction (PPI) network of each of the four sets of DEGs was constructed using the STRING website v11.0 (https://www.string-db.org/) and exported to Cytoscape software v3.7.1 for visualisation [20]. Molecular Complex Detection (MCODE) plugin in Cytoscape was used to identify clusters that were highly interconnected in a network [21]; the top three scored sub-networks were then subjected to GO analysis.
Gene set enrichment analysis of DEGs
Gene set enrichment analysis (GSEA) was used to ascertain the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for the DEGs identified in each of the four comparisons. The normalised enrichment score (NES) was used to quantify the enrichment magnitude [22, 23].
Participants and ethical approval
Participants comprising COPD patients and healthy participants (controls) were recruited from the China–Japan Friendship Hospital. COPD patients were diagnosed based on the GOLD clinical criteria. Lung tissue samples were collected from the COPD patients and controls scheduled for pulmonary lobectomy. The lung function data and basic information of the participants are shown in table 2.
Clinical characteristics of enrolled subjects from whom lung tissues were collected
All experimental work with humans was approved by the Ethics Committee of China–Japan Friendship Hospital. All participants provided written informed consent.
Cell culture and treatment with cigarette smoke extract
The murine lung epithelial (MLE) cell line MLE-12 was maintained in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Cigarette smoke extract (CSE) was prepared by a modification of a previously published method [24]. Briefly, one 3R4F reference cigarette containing 9.4 mg tar and 0.73 mg nicotine (University of Kentucky, USA) was bubbled into 10 mL of high-glucose DMEM (Gibco), and it was subsequently diluted to 5% in use. The MLE-12 cells were then treated with 5% CSE for 24 h.
Immunohistochemical analysis
Lung tissues were fixed in 4% paraformaldehyde solution (Beyotime, Nantong, China) for 24 h and then dehydrated, embedded in paraffin and sectioned following routine methods. The sections were incubated with anti-HIF-3α antibody (1:100 dilution; Proteintech, Wuhan, China) overnight at 4°C, followed by incubating with anti-horseradish peroxidase (HRP) antibody (1:200 dilution, SeraCare, Beijing, China) at room temperature for 30 min. 3,3'-diaminobenzidine (DAB) was used for colour development. The results were scored as the integrated optical density/area as detected by Image-Pro Plus.
Protein lysis and Western blotting
Equal amounts of protein (20 μg) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA, USA). After 1 h of blocking with 10% milk, the membranes were incubated with anti-HIF-3α (ABclonal, Wuhan, China) or anti-GAPDH (ABclonal) antibody at 4°C overnight. After incubation with HRP-conjugated secondary antibody (ABclonal) for 1 h, protein bands were developed in an automatic exposure machine (ProteinSimple, Minneapolis, MN, USA).
The reactive oxygen species measurement
MLE-12 cells were incubated with 10 μM DCFH-DA (Solarbio, Beijing, China) at 37°C for 30 min. The supernatant was removed, and the cells were washed with DMEM. Images viewed under a fluorescence microscope were obtained.
Superoxide dismutase assay
The relative superoxide dismutase (SOD) concentration in cell lysates was assessed using a SOD assay kit (Beyotime, Nantong, China) according to the manufacturer's instructions.
CCK-8 assay
A CCK-8 assay was performed to detect cell viability according to the manufacturer's protocol (Beyotime). ∼5×103 cells were seeded in 96-well plates. The cells were treated with 10 μL of CCK-8 solution for 24 h and incubated in the dark for another 2 h. The absorbance was measured at a wavelength of 450 nm.
Statistical analysis
All experiments were performed independently at least three times. The data are presented as mean±standard error of the mean (sem). Comparisons between the two groups were performed using two-tailed t-tests. p<0.05 was considered statistically significant.
Results
Identification of DEGs during the progression of COPD
We identified DEGs (adjusted p<0.05 and |log2fold change| ≥0.5) in each of the four datasets. There were 577 upregulated genes and 482 downregulated genes in the GSE37768 dataset (smokers versus nonsmokers), 1832 upregulated genes and 1798 downregulated genes in the GSE119040 dataset (emphysema versus non-emphysema patients), 1757 upregulated genes and 2851 downregulated genes in the GSE1650 dataset (severe versus mild emphysema patients), and 654 upregulated genes and 998 downregulated genes in the GSE69818 dataset (GOLD 4 versus GOLD 1 COPD patients).
GO analyses of DEGs
Regarding the GO analysis of the GSE37768 dataset (smokers versus nonsmokers), the DEGs were mainly enriched in “negative regulation of apoptotic process”, “immune response” and “inflammatory response”, functioned in “nuclear” and “perinuclear of cytoplasm” through “protein binding” and “receptor activity” (figure 1a) (supplementary figure S1a). Regarding the GSE119040 dataset (emphysema versus non-emphysema patients), the DEGs were mainly enriched in “negative regulation of the apoptotic process”, “innate immune response” and “extracellular matrix organisation”, functioning in “cytoplasm” through “protein binding” (figure 1b) (supplementary figure S1b). Regarding the GSE1650 dataset (severe versus mild emphysema patients), the DEGs were mainly enriched in “apoptotic process”, “oxidation–reduction process”, “protein phosphorylation” and “inflammatory response”, functioning in “cytoplasm” through “protein binding” (figure 1c) (supplementary figure S1c). Lastly, regarding the GSE69818 dataset (GOLD 4 versus GOLD 1 patients), the DEGs were mainly enriched in “immune response”, “inflammatory response” and “oxidation–reduction process”, functioning in the “extracellular exosome” through “protein binding” (figure 1d) (supplementary figure S1d). The cellular component results show, in the progression of COPD, the location where DEG functions shift from the nuclear and perinuclear at the beginning, to the cytoplasm and finally to the extracellular exosome.

Gene ontology (GO) enrichment analyses for upregulated genes. GO enrichment analyses for: a) smokers versus nonsmokers; b) emphysema versus non-emphysema patients; c) severe versus mild emphysema patients; and d) Global Initiative for Chronic Obstructive Lung Disease (GOLD) 4 versus GOLD 1 patients. BP: biological process; CC: cellular component; MF: molecular function.
GO analyses of PPI networks
To understand the relationships among the four sets of DEGs at the protein level, i.e. the relationships among the differentially expressed proteins (DEPs), and to analyse their functions in detail, we constructed four PPI networks. MCODE was applied for further analysis, and the top three densely connected sub-networks in each PPI network were identified, and then GO analyses were conducted for each of them [21].
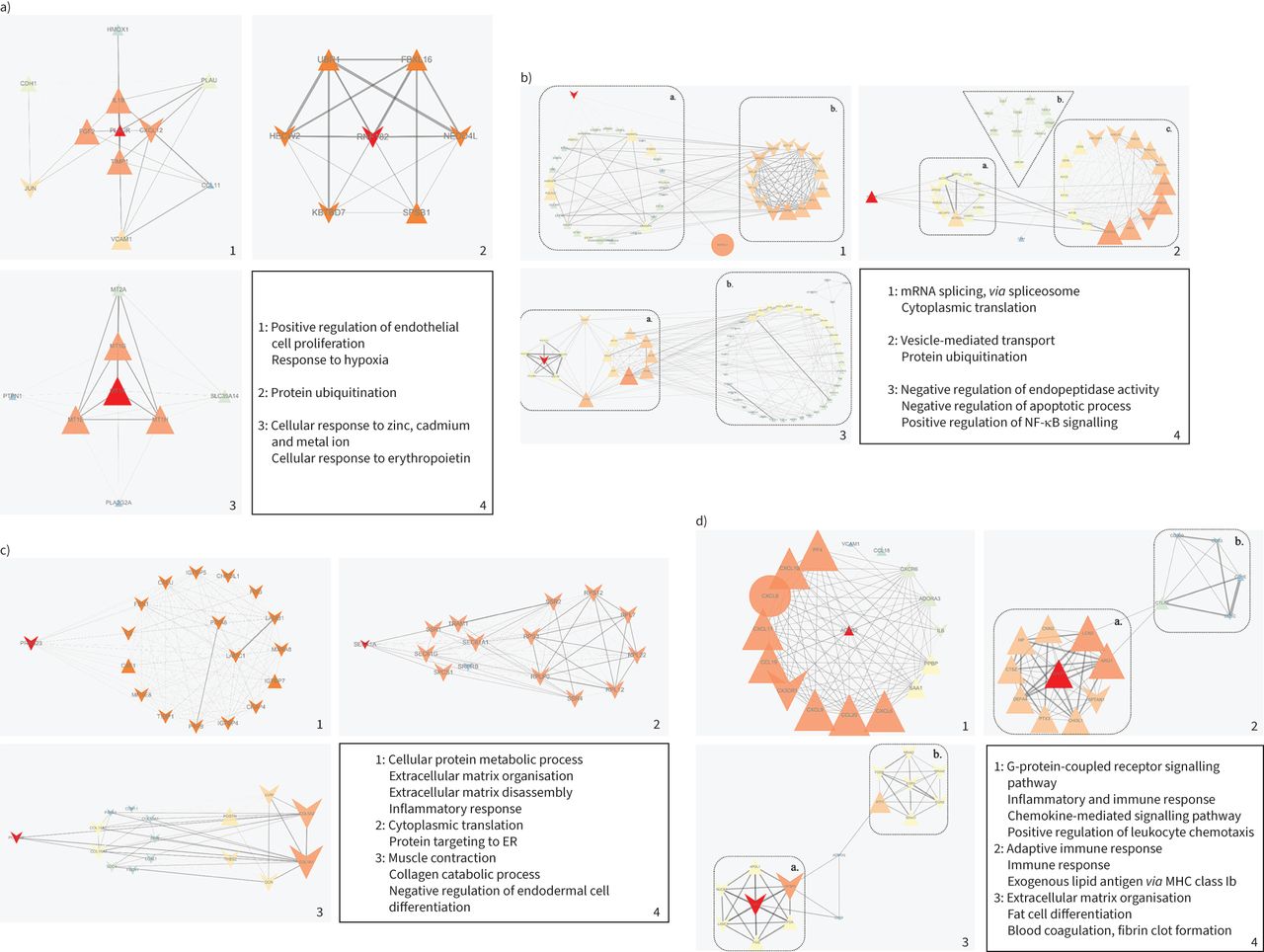
Regarding smoking versus non-smoking, the functions of the three sub-networks were mainly enriched in “positive regulation of endothelial cell proliferation”, and “cellular response to zinc, cadmium, metal ions and erythropoietin” (figure 2a). In the progression to emphysema, the functions of the three sub-networks were mainly enriched in “cytoplasmic translation”, “negative regulation of apoptotic process” and “positive regulation of NF-κB signalling” (figure 2b). To further explore the functions of the sub-networks, we analysed the sub-networks in parts. The first sub-network consisted of parts a and b (figure 2b.1). The enriched functions of part a included “mRNA splicing, via spliceosome”. The enriched functions of part b included “nuclear-transcribed mRNA catabolic process” and “nonsense-mediated decay”. Parts a and b interacted through NHP2L1. The second sub-network consisted of parts a, b and c (figure 2b.2). The enriched functions of part a included “ephrin receptor signalling pathway”. The enriched functions of part b included “protein ubiquitination”. The enriched functions of part c included “retrograde vesicle-mediated transport, Golgi to ER”. Parts a and c interacted through UBE2N. The third sub-network consisted of parts a and b (figure 2b.3). The enriched functions of part a included “mitochondrial electron transport” and “ubiquinol to cytochrome c”. The enriched functions of part b included “interferon-γ-mediated signalling pathway”.

Protein–protein interaction (PPI) networks of differentially expressed proteins (DEPs). Graphs 1, 2 and 3 show the top three sub-networks of differentially expressed genes (DEGs) and graph 4 shows the enriched gene ontology (GO) biological process (BP) terms for a) smoking versus non-smoking patients, b) emphysema versus non-emphysema patients, c) severe versus mild emphysema patients and d) Global Initiative for Chronic Obstructive Lung Disease (GOLD) 4 versus GOLD 1 COPD patients. Triangles represent upregulated DEPs, “V” shapes represent downregulated DEPs and circles represent proteins produced by the string database, which have no state in our data. Larger node size and a more orange colour indicate higher degree value. Thicker edges represent stronger interactions (higher combined score of DEPs). NF-κB: nuclear factor kappa B; ER: endoplasmic reticulum; MHC: major histocompatibility complex.
Regarding the progression from mild to severe emphysema, the functions of the three sub-networks were mainly enriched in “extracellular matrix organisation”, “extracellular matrix disassembly”, “inflammatory response”, and “negative regulation of endodermal cell differentiation” (figure 2c). Regarding the progression from GOLD 1 to GOLD 4, the functions of the three sub-networks were mainly enriched in “inflammatory and immune response”, “positive regulation of leukocyte chemotaxis”, “fat cell differentiation” and “blood coagulation, fibrin clot formation” (figure 2d). The second sub-network consisted of parts a and b (figure 2d.2). The enriched functions of part a included “innate immune response” and “cellular response to hydrogen peroxide”. The enriched functions of part b included “adaptive immune response”. The third sub-network consisted of parts a and b (figure 2d.3). The enriched functions of part a included “positive regulation of RNA polymerase II promoter transcription” and “fat cell differentiation”. The enriched functions of part b included “extracellular matrix organisation” and “positive regulation of ERK1 and ERK2 cascade”.
Along with the GO analysis above, the results demonstrated a persistently enhanced oxidation–reduction process, and immune and inflammatory response in the development of COPD. Besides, in smoker versus nonsmoker and emphysema versus non-emphysema patients, it showed a negative regulation of apoptotic process. Interestingly, in severe versus mild emphysema patients and GOLD 4 versus GOLD 1 patients, it showed a positive regulation of apoptotic process and intensified immune and inflammatory responses, suggesting that there may be a compensation threshold in the development of COPD. When the damage reaches the threshold, the organism exhibits an injury response, which may be related to the lung inflammation and persists even after smoking cessation in patients with COPD [1].
Identification of core DEGs
Taken together, the GO and PPI network analyses indicated that continuously immune and inflammatory responses and oxidative stress are exhibited in the development of COPD. We further identified and annotated 14 DEGs that were consistently either upregulated or downregulated in the four comparisons (supplementary tables S1–4), six of which were upregulated (figure 3a) (C10orf10, MSMB, IGLV1-44, CYP1B1, ZNF385D and MUC4) and eight of which were downregulated (figure 3b) (PPFIBP1, DDX17, FOSB, SVEP1, RABGAP1L, HIF3A, PTCH1 and PTPRD). By annotating these 14 DEGs, we found that they were associated with the comorbidities of COPD, such as anxiety and depression, atherosclerosis and nonsmall cell lung cancer (NSCLC), which would provide evidence for future in-depth studies on the comorbidities of COPD (supplementary tables S3 and S4).

Venn and logistic regression analysis. Venn diagrams showing a) six consistently upregulated differentially expressed genes (DEGs) and b) eight consistently downregulated DEGs. Receiver operating characteristic (ROC) curve analysis of the core genes prediction model. c) Construction of the logistic prediction model. d) Validation of the logistic prediction model. AUC: area under the curve.
Logistic regression prediction model based on the core DEGs
A logistic regression prediction model based on the core DEGs was constructed to distinguish between COPD patients and non-COPD patients, using GSE37768 data as the training set and GSE8581 data as the test set. The AUC for the training set was 0.828 (figure 3c), while the sensitivity and specificity were 71.7% and 80%, respectively. The AUC for the test set was 0.750 (figure 3d), while the sensitivity and specificity were 74.1% and 60.2%, respectively. The results above demonstrated the core DEGs were not only meaningful in biological analysis, but also have efficacy in distinguishing COPD patients from non-COPD patients in clinical practice.
GSEA of DEGs
We performed a GSEA of the four sets of DEGs in order to identify core pathways that 1) were consistently upregulated or downregulated in all four comparisons and 2) had a NES ≥1 in at least three of the comparisons. We identified five core pathways; the following two pathways were continuously upregulated: “alanine aspartate and glutamate metabolism” and “pathogenic Escherichia coli infection”, while the following three pathways were continuously downregulated: “phosphatidylinositol signalling system”, “oocyte meiosis” and “ubiquitin-mediated proteolysis” (figure 4b) (supplementary figure S2).
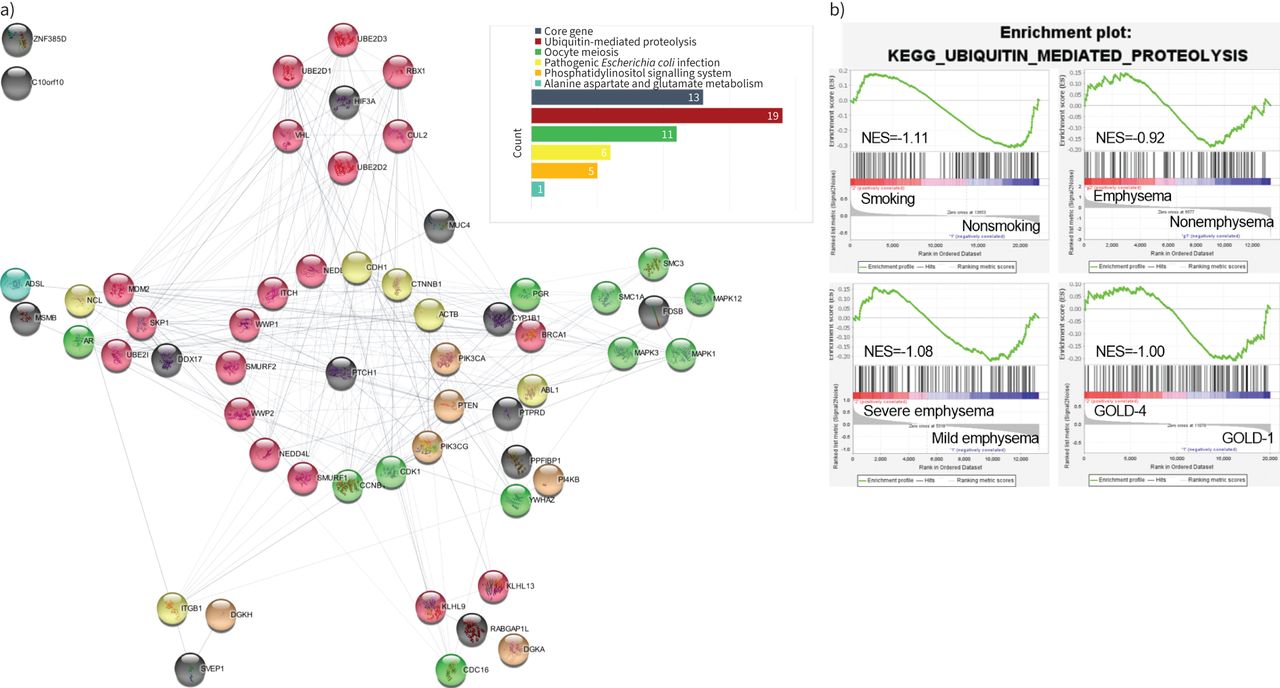
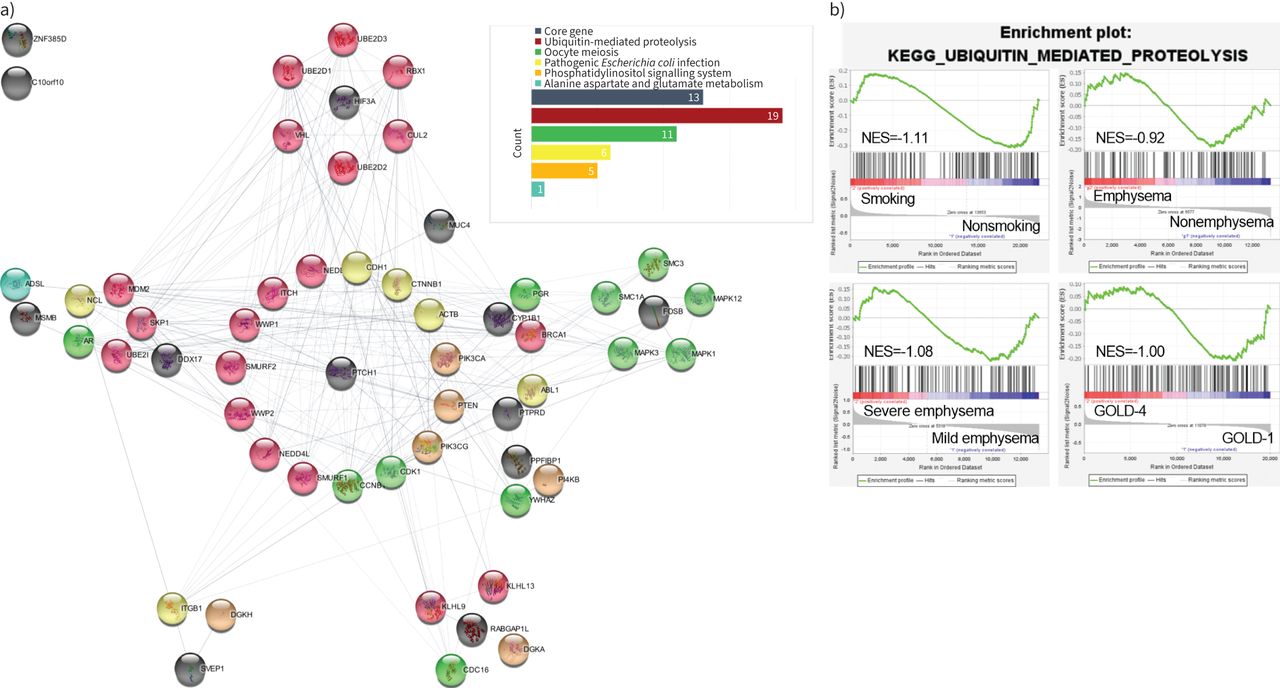
Protein–protein interaction (PPI) network of consistently upregulated or downregulated differentially expressed proteins (DEPs) and consistently enriched pathways. a) Black solid circles indicate that the protein is encoded by a core differentially expressed gene (DEG). Red, green, yellow, orange and blue solid circles indicate that the protein belongs to the ubiquitin-mediated proteolysis pathway (continuously downregulated), oocyte meiosis pathway (continuously downregulated), pathogenic Escherichia coli infection pathway (continuously upregulated), phosphatidylinositol signalling system pathway (continuously downregulated) and alanine aspartate and glutamate metabolism pathway (continuously upregulated), respectively. b) The co-downregulation pathway in strongest interaction with core DEPs.
PPI network of core DEPs and core pathways
To investigate the interactions between the five core pathways and 14 core DEPs, we constructed a PPI network (the flow of the construction is shown in supplementary figure S3). The results indicated that 42 proteins out of a total of 394 proteins in the five pathways interacted with 11 of the core DEPs (figure 4a). Among the 42 proteins, there were 19 proteins belonging to the ubiquitin-mediated proteolysis pathway, followed by 11, six, five and one belonging to the oocyte meiosis, pathogenic Escherichia coli infection, phosphatidylinositol signalling system, and alanine aspartate and glutamate metabolism pathways, respectively (supplementary table S5). Among the 14 core DEGs, HIF3A exhibited specialised interactions with six proteins that all belonged to the ubiquitin-mediated proteolysis pathway, further suggesting the interaction between HIF3A and ubiquitin-mediated proteolysis pathway may play a key role in the progression of COPD.
Validation of HIF-3α in vitro and in vivo
To validate our findings, we evaluated HIF-3α expression by immunohistochemical analysis and Western blotting of lung tissue from COPD patients and healthy participants. Immunohistochemical analysis showed that HIF-3α protein expression (concentrated in alveolar epithelial cells) was decreased in COPD (figure 5a and b). Western blotting confirmed the HIF-3α protein downregulation in COPD (figure 5c and d). We also established a COPD cell model, and Western blotting confirmed that HIF-3α protein expression was downregulated in CSE-exposed MLE-12 cells (figure 5e and f). These results indicate downregulation of HIF-3α expression in COPD tissues in vivo and CSE-exposed MLE-12 cells in vitro.

HIF-3α is reduced in the lungs of COPD patients and cigarette smoke extract (CSE)-treated murine lung epithelial (MLE)-12 cells. a) Immunohistochemistry showed that HIF-3α was markedly lower in COPD patients than controls. Original magnification ×40. Scale bars: 20 μm. b) Immunohistochemical (IHC) score (mean±sem) in airway epithelial cells based on 10 image fields (40×) per sample. c, d) Western blotting of human lung homogenates from COPD and control patients that were probed with an anti-HIF-3α antibody (normalised to GAPDH). e, f) Western blotting of CSE-exposed MLE-12 cells (exposed for 24 h) that were probed with an anti-HIF-3α antibody (normalised to GAPDH). IOD: integrated optical density. **: p<0.01; ***: p<0.005; ****: p<0.001.
The HIF3A overexpression mitigates CSE-induced reactive oxygen species accumulation
Oxidative stress arises as a result of endogenous antioxidant defences being genetically impaired and/or overwhelmed by the presence of reactive oxygen species (ROS) [25]. SOD plays an important role in antioxidant activity and prevents ROS-initiated reactions [26]. To further explore the interactions between HIF3A and oxidative stress, a lentivirus vector was used to promote HIF3A overexpression in CSE-treated MLE-12 cells. Overexpression of HIF3A (figure 6a and b) decreased ROS production (figure 6e) and restored SOD levels (figure 6c). Besides, the low cell viability (figure 6d) caused by CSE was alleviated after the HIF3A overexpression. These results suggest that the HIF3A overexpression mitigated oxidative stress and further enhanced cell viability.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HIF3A overexpression mitigates cigarette smoke extract (CSE)-induced reactive oxygen species (ROS) accumulation and the decreased cell viability in murine lung epithelial (MLE)-12 cells. a,b) MLE-12 cells were transfected with HIF3A overexpression (OE) vector (Lenti-shHIF3A) or control vector (Lenti-Ctrl). HIF-3α protein levels were detected by Western blot analysis and normalised to GAPDH levels. c) Superoxide dismutase (SOD) levels in each group. d) Cell viability of each group was analysed by the CCK-8 assay and all values were normalised to those of the control group. e) Images of intracellular ROS stained by DCFH-DA were captured with a fluorescence microscope. Scale bars: 20 μm. The results were presented as the mean±sem for three independent experiments. *: p<0.05; **: p<0.01; ns: p>0.05 (nonsignificant).
Discussion
As late diagnosis of COPD is a major factor in the poor prognosis of many patients [7–9], research on COPD should be shifted to the period of pathological changes rather than the period of clinical symptoms. In this study, we conducted a GET analysis and divided COPD development into four developmental stages based on aetiology, pathology and the worsening of clinical symptoms (exposure to smoking, from non-emphysema to emphysema, from mild to severe emphysema, and from GOLD 1 to GOLD 4) and analysed GEO microarray datasets related to these stages, carrying out a holistic analysis by integrating these four datasets to uncover the gene function characteristics in each of the progression stages and unveiling the underlying mechanisms.
GET analysis is a novel approach, and it may lead to improved understanding of underlying pathogenic mechanisms and identification of novel targets [10]. By conducting a GET analysis in this study, we revealed that, during the progression of COPD, immune and inflammatory response was continuously enriched, accompanied by intensified oxidation-reduction process, positive regulation of apoptotic and leukocyte chemotaxis, and cellular response to hydrogen peroxide. These functions were especially intensified from mild to severe emphysema and from GOLD 1 to GOLD 4. This suggests that patients exhibit persistently increased inflammation and oxidative stress after the emphysema stage develops, which further highlights, in clinical practice, special attention should be paid to identifying emphysema and to monitoring and controlling oxidative stress, which provides more precise insights into the GOLD report.
Given the sustained progression of oxidative stress and sustained immune and inflammatory responses, we identified 14 core DEGs that were continuously upregulated or downregulated in all four comparisons. Of these, six were continuously upregulated (C10orf10, MSMB, IGLV1-44, CYP1B1, ZNF385D and MUC4) and eight were continuously downregulated (PPPFIBP1, DDX17, FOSB, SVEP1, RABGAP1L, HIF3A, PTCH1 and PTPRD). These core DEGs were associated with the comorbidities of COPD such as anxiety and depression, atherosclerosis and NSCLC (supplementary tables S3 and S4). The results above further support the idea that COPD is a systemic syndrome rather than a uniform disease entity [27, 28].
In addition, to identify the potential pathways involved in the development of COPD, we performed GSEA on each of the four datasets and identified five core pathways. Two of them were continuously upregulated (“alanine aspartate and glutamate metabolism”, “pathogenic Escherichia coli infection”), and three of them were continuously downregulated (“oocyte meiosis”, “phosphatidylinositol signalling system” and “ubiquitin-mediated proteolysis”).
To elucidate the potential mechanisms underlying the development of COPD, the proteins encoded by the core DEGs and the enriched core pathways were used to construct a PPI network. Among the core DEPs, HIF-3α attracted our attention, as it exhibited specialised interactions with six proteins that all belonged to the ubiquitin-mediated proteolysis pathway. HIF3A is a member of the hypoxia-inducible transcription factor (HIF) family, which are master regulators of the adaptive cell response to decreased oxygen levels, controlling the expression of many genes in an oxygen-dependent manner; the product of these genes was involved in haematopoiesis, angiogenesis, iron transport, oxidation stress and extracellular matrix synthesis [29]. There are three members in this family: HIF1A, HIF2A (EPAS1) and HIF3A. Among them, HIF3A is the most recently identified member. Compared with HIF-1α and HIF-2α, HIF-3α has dual functions: inhibition of the activities of HIF-1α and HIF-2α, and regulation of its own target genes in a response to hypoxia [30, 31]. It can upregulate genes involved in glucose and amino acid metabolism, apoptosis, proteolysis, p53 signalling, PPAR signalling, Jak-STAT signalling and NOD-like receptor signalling [31]. In addition, it can inhibit the production of ROS and reduce oxidative stress levels [31–34]. Therefore, decreased HIF3A may be a major factor that causes and aggravates the oxidative stress in COPD.
In the PPI analysis of core DEPs and core pathways, the interaction between HIF3A and von Hippel–Lindau (VHL) attracted our attention (figure 4). Under normal oxygen partial pressure, HIF-3α can be degraded by the pVHL ubiquitin–proteasome due to the shared common oxygen-dependent degradation (ODD) domain in HIF-3α. The ability of VHL to degrade HIF-3α is dependent on the proline 490 residue of HIF-3α, and this is increased in the presence of prolyl hydroxylase (PHD), a cellular sensor for low oxygen [35]. PHD catalyses the hydroxylation of key amino acid residues in the HIF-α ODD domain [36, 37]. This is followed by VHL binding to HIF-α and inducing degradation via the ubiquitin-proteasome pathway [38, 39]. A previous study showed that HIF-3α expression was elevated during acute hypoxia and decreased slowly after 14 days of hypoxia, accompanied by sustained HIF-1α and HIF-2α upregulation [40, 41]. Combined with these results, the transient elevated HIF-3α may be due to the inhibited PHD and VHL under hypoxia, and in chronic hypoxia (which often occurs in COPD), HIF-3α downregulation may be due to the competition with HIF-1α and HIF-2α in binding to the HIF-1β subunits [42]. In vitro and in vivo experiments showed that under hypoxia HIF-1α and HIF-2α upregulation can lead to ROS accumulation [32], and HIF1A and HIF2A knockdown reduces the oxidative stress [33, 34]. According to the 2022 GOLD report [1], “oxidative stress may be an important amplifying mechanism in COPD”. Oxidative stress is an imbalance between oxidants and antioxidants in favour of the oxidants, leading to a disruption of redox signalling and control and/or molecular damage [43], and as HIF3A has transcriptional activation ability under hypoxia and is a negative regulator of HIF1A and HIF2A [31, 42], the downregulated HIF3A in our study may be one of the reasons for the persistent elevated oxidative stress level and the amplifying mechanism in COPD.
Another of the core DEPs, the PTCH1 (patched 1) protein, also attracted our attention. According to the results of a genome-wide association study on COPD, PTCH1 showed the strongest positive associations with FEV1/FVC [44]. PTCH1 encodes a member of the patched family of proteins and a component of the hedgehog signalling pathway. Similarly, the 2022 GOLD report also indicated that HHIP, which is part of the same hedgehog signalling pathway as PTCH1, is highly correlated with COPD phenotype, but the exact pathogenic mechanism remains unclear, suggesting that our analytical approach can indeed identify key factors in the pathogenesis of COPD.
In this study, we applied a GETomics approach to conduct a holistic analysis of COPD, and identified that oxidative stress was especially intensified in mild to severe emphysema and from stages GOLD 1 to GOLD 4, which provides more precise insight into the GOLD report and clinical practice. According to the continuously intensified oxidative stress, and immune and inflammatory response, we identified and annotated 14 core DEGs that were consistently either upregulated or downregulated in the four comparisons, and found these core DEGs were related to the comorbidities of COPD. By constructing and validating the prediction model, we discovered these 14 core DEGs were not only meaningful in biological analysis, but also have efficacy in differentiating COPD patients from healthy individuals. Through further GSEA and PPI analysis, we found the highly interacting HIF3A and ubiquitin-mediated proteolysis pathway may contribute to the oxidative stress in COPD pathogenesis. Finally, a lentivirus was used to promote HIF3A overexpression, and the results suggest overexpression of HIF3A mitigates oxidative stress and further enhances cell viability in CSE-treated MLE-12 cells. Two earlier studies have reported on HIF3A expression in COPD, one of which demonstrated that HIF3A mRNA was downregulated in the tibialis anterior muscle of severe COPD patients, which was consistent with our study [45]. However, another study reported that HIF-3α was upregulated in the lungs of mice who were exposed to cigarette smoke for 15 weeks [46]. As HIF3A is the most recently identified member of the HIF family, its specific regulatory mechanisms need further research.
The current study has some limitations. We only assessed HIF3A expression in lung and MLE-12 cells, so its expression in serum and sputum remains unknown. Serum and sputum are more easily assessed and therefore more widely used in clinical practice to assess biomarkers. We intend to study HIF3A expression in serum and sputum in the future.
Conclusion
In conclusion, the results of the GET analysis strongly indicate that special attention should be paid by clinicians to identifying emphysema and monitoring and controlling COPD patients’ oxidative stress levels. Besides, the 14 core DEGs have efficacy in differentiating COPD patients from healthy individuals and may be related to the comorbidities. Furthermore, overexpression of HIF3A can alleviate oxidative stress in COPD.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00343-2022.SUPPLEMENT
Footnotes
Provenance: Submitted article, peer reviewed.
Data availability statement: Publicly available datasets were analysed in this study. These data can be found in the GEO database, accession numbers GSE37768, GSE119040, GSE1650, GSE69818 and GSE8581.
Ethics approval and consent to participate: This study was approved by the Ethics Committee of China–Japan Friendship Hospital.
Author contributions: J. Jiang, S. Chen and T. Yang participated and conceived the study design. J. Jiang, T. Yu and C. Chang collected the data. J. Jiang performed the experiments, analysed the data and wrote the manuscript. J. Jiang, J. Liu, X. Ren, C. Wang and T. Yang interpreted and discussed the data. C. Wang, T. Yang, K. Huang, H. Niu and B. Li refined the final draft and revised the manuscript. All the authors reviewed the final version of the manuscript.
Conflict of interest: J. Jiang has nothing to disclose.
Conflict of interest: S. Chen has nothing to disclose.
Conflict of interest: T. Yu has nothing to disclose.
Conflict of interest: C. Chang has nothing to disclose.
Conflict of interest: J. Liu has nothing to disclose.
Conflict of interest: X. Ren has nothing to disclose.
Conflict of interest: H. Niu has nothing to disclose.
Conflict of interest: K. Huang has nothing to disclose.
Conflict of interest: B. Li has nothing to disclose.
Conflict of interest: T. Yang has nothing to disclose.
Conflict of interest: C. Wang has nothing to disclose.
Support statement: This study was funded by a grant from CAMS Innovation Fund for Medical Science (2021-I2M-1-049) and National High Level Hospital Clinical Research Funding (2022-NHLHCRF-LX-01-01-01). The sponsors had no role in any of the stages from study design to submission of the manuscript for publication. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received July 12, 2022.
- Accepted October 6, 2022.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










