





Abstract
Background Oral imatinib has been shown to be effective, but poorly tolerated, in patients with advanced pulmonary arterial hypertension (PAH). To maintain efficacy while improving tolerability, AV-101, a dry powder inhaled formulation of imatinib, was developed to deliver imatinib directly to the lungs.
Methods This phase 1, placebo-controlled, randomised single ascending dose (SAD) and multiple ascending dose (MAD) study evaluated the safety/tolerability and pharmacokinetics of AV-101 in healthy adults. The SAD study included five AV-101 cohorts (1 mg, 3 mg, 10 mg, 30 mg, 90 mg) and placebo, and a single-dose oral imatinib 400-mg cohort. The MAD study included three AV-101 cohorts (10 mg, 30 mg, 90 mg) and placebo; dosing occurred twice daily for 7 days.
Results 82 participants (SAD n=48, MAD n=34) were enrolled. For the SAD study, peak plasma concentrations of imatinib occurred within 3 h of dosing with lower systemic exposure compared to oral imatinib (p<0.001). For the MAD study, systemic exposure of imatinib was higher after multiple doses of AV-101 compared to a single dose, but steady-state plasma concentrations were lower for the highest AV-101 cohort (90 mg) compared to simulated steady-state oral imatinib at day 7 (p=0.0002). Across AV-101 MAD dose cohorts, the most common treatment-emergent adverse events were cough (n=7, 27%) and headache (n=4, 15%).
Conclusions AV-101 was well tolerated in healthy adults, and targeted doses of AV-101 significantly reduced the systemic exposure of imatinib compared with oral imatinib. An ongoing phase 2b/phase 3 study (IMPAHCT; clinicaltrials.gov identifier NCT05036135) will evaluate the safety/tolerability and clinical benefit of AV-101 for PAH.
Abstract
AV-101, a dry-powder inhaled formulation of imatinib, reduces systemic exposure to imatinib versus oral imatinib and is well tolerated in healthy adults. An ongoing phase 2b/3 study will evaluate AV-101 in patients with pulmonary arterial hypertension. https://bit.ly/3Tlh6ru
Introduction
Pulmonary arterial hypertension (PAH) is a rare, life-limiting disease characterised by excessive pulmonary vasoconstriction and abnormal vascular remodelling [1–3], including hyperproliferation of fibroblasts, smooth muscle cells and endothelial cells in the pulmonary vasculature [4, 5]. Approved therapies for PAH management focus on the resultant vasoconstrictive pathophysiology to improve haemodynamics and exercise tolerance. These therapies do slow disease progression [6], but PAH continues to be associated with high morbidity and mortality; median survival after diagnosis is ∼7 years in contemporary registries [7–9].
Imatinib is a small-molecule kinase inhibitor, initially approved for the treatment of chronic myeloid leukaemia [10], which inhibits tyrosine kinases involved in growth, differentiation, proliferation, survival, inflammation, metabolism and apoptosis [10–12]. Imatinib most potently inhibits platelet-derived growth factor receptor (PDGFR), discoidin domain receptor (DDR), KIT proto-oncogene receptor tyrosine kinase (KIT), colony stimulating factor 1 receptor (CSF1R) and abelson murine leukaemia viral oncogene homologue (ABL) kinases [13]. Signalling through these kinases has been implicated (directly and indirectly) in PAH remodelling, including PDGFR-mediated proliferation and resistance to apoptosis of vascular smooth muscle cells [14, 15], KIT expression in the vasculature and its influence on precursor cells [16], fibrotic signalling and recovery mediated by DDR [17] and ABL [18] and immune dysregulation via CSF1R and KIT [16, 19].
Imatinib's antiproliferative and pro-apoptotic properties have been demonstrated in multiple pre-clinical studies; in in vitro and in vivo PAH models, imatinib reversed aspects of pulmonary vascular remodelling, attenuated right heart hypertrophy and right ventricular pressure, improved haemodynamics and reduced vascular smooth muscle cell proliferation and neointima formation [14, 15, 20–22]. In a phase 2 study conducted by Novartis, patients with PAH treated with oral imatinib demonstrated a significant reduction in pulmonary vascular resistance [23]. When assessed in a phase 3 randomised controlled trial (IMPRES), oral imatinib (400 mg) treatment resulted in significant improvements in 6-min walk distance and haemodynamics observed in patients on two or more background PAH therapies with persistent symptoms [24, 25]. However, patients treated with oral imatinib in the IMPRES study had high discontinuation rates and adverse events, particularly an excess of subdural haematoma in patients taking concomitant vitamin K antagonists [24]. After the IMPRES study, Novartis chose not to pursue the development of oral imatinib for PAH.
AV-101 is an inhaled dry powder formulation of imatinib developed to deliver effective concentrations of imatinib to respiratory tissue while simultaneously limiting systemic exposure and potentially circumventing systemic adverse events associated with oral imatinib. The objective of the current study was to characterise the pharmacokinetics and safety/tolerability of AV-101 in healthy adult participants.
Methods
Study design and participants
This was a phase 1, single-centre, placebo-controlled, randomised study that evaluated the pharmacokinetics and safety/tolerability of single ascending doses (SADs) and multiple ascending doses (MADs) of AV-101. Eligible participants were healthy adults, aged 18–59 years, with no clinically significant medical conditions, a body mass index of 18.0–35.0 kg·m−2 and negative tests for coronavirus disease 2019, HIV, hepatitis B and hepatitis C. Additional details on the study design and participants are described in the supplementary methods. The primary safety outcomes were based on medical, physical, laboratory and treatment-emergent adverse event (TEAE) evaluations.
This trial was conducted in accordance with the Declaration of Helsinki and good clinical practice, and the study protocol was approved by the independent ethics committee/institutional review board at the study centre. All participants provided written informed consent.
Selection of doses
AV-101 dosing was based on a weight-of-evidence approach from the results of pre-clinical studies and pharmacokinetic modelling, with the goal of achieving substantially lower systemic exposure versus a 400-mg oral dose of imatinib. Imatinib has a demonstrated bioavailability of 98% and a peak plasma concentration (Cmax) that is achieved within 2–4 h post-dose [26]. A substantial dose reduction was expected using inhalation as the route of administration since ≥10-fold dose reductions have been observed for inhaled drugs relative to their oral counterparts, even after accounting for bioavailability (e.g. albuterol, treprostinil) [27, 28]. The 1-mg starting dose in the first SAD cohort was <1/100 of the dose of oral imatinib administered to patients with PAH in prior clinical trials and was therefore considered a conservative starting dose of AV-101.
The AV-101 capsules were of two dose strengths: 1 mg and 10 mg. For each dosing time point, participants inhaled from one capsule in the 1-mg and 10-mg cohorts, three capsules in the 3-mg and 30-mg cohorts and nine capsules in the 90-mg cohorts.
SAD study
The SAD study included five AV-101 cohorts (1 mg, 3 mg, 10 mg, 30 mg, 90 mg), with eight participants randomised to AV-101 (n=6) or placebo (n=2) per cohort. Study treatment was administered in the morning after food by oral inhalation delivered via a dry powder inhaler. One additional cohort of eight participants received a nonblinded, single dose of oral imatinib 400 mg with food and ∼240 mL of water. The first dose in a cohort was administered ≥5 days after dosing in the previous cohort to allow for assessment of safety/tolerability.
MAD study
The MAD study involved three AV-101 cohorts (10 mg, 30 mg, 90 mg), with up to 12 participants randomised to AV-101 (n=9) or placebo (n=3) per cohort, with treatment administered twice daily for 7 days, except for a single morning dose only on day 7 (figure 1). Doses of AV-101 for the MAD cohorts were determined based on results of the SAD study, with a 10-mg twice daily starting dose and a 90-mg twice daily maximum dose. Morning doses were administered ∼30 min after food, and evening doses were administered ∼12 h later but at any time after food. The first dose in a cohort was administered ≥10 days after the first dose in the previous cohort to allow for assessment of safety/tolerability.

Multiple ascending dose (MAD) study design and schedule of assessments. EOT: end of treatment; EOS: end of study; PK: pharmacokinetics; SpO2: oxygen saturation. #: the morning dose of AV-101 or placebo was administered within 30 min of food, and the evening dose of AV-101 was administered ∼12 h later; ¶: PK blood samples were taken on days 1 and 7 prior to and after the morning dose at 5, 20 and 40 min and at 1, 2, 4, 6, 9 and 12 h. On days 2–6, blood samples for pharmacokinetic evaluation were taken prior to dosing only. Additional samples were taken at 24, 48 and 72 h after the final dose (i.e. on days 8, 9 and 10); +: spirometry assessments (per American Thoracic Society guidelines) were performed at screening, on day 0, prior to dosing and at 20 min, 60 min and 4 h after the morning dose on days 1, 3 and 7 and at the follow-up visit on day 14; §: vital signs (systolic and diastolic blood pressure, heart rate and respiratory rate) and SpO2 were recorded at screening, on day 0, prior to each dose, at 1 h after each dose, the morning of discharge on day 10 and the follow-up visit on day 14; ƒ: laboratory tests (haematology, biochemistry and urinalysis) were performed at screening, on days 0–3, 7 and 8 and at the follow-up visit on day 14.
Pharmacokinetics
Plasma concentrations of imatinib and its major metabolite, N-desmethyl imatinib, were measured to establish their pharmacokinetic profile after AV-101 inhalation in both SAD and MAD portions, together with the plasma concentrations of imatinib following the single oral imatinib 400-mg dose administered in the SAD study. Noncompartmental pharmacokinetic analysis of the plasma concentration-versus-time profiles were performed using Phoenix WinNonlin (Certara) software, version 8.0 or higher.
In the SAD study, blood samples were taken prior to dosing and after dosing at 5, 20 and 40 min and at 1, 2, 4, 6, 9, 12, 48 and 72 h post-dose.
For oral imatinib in the MAD study, a population pharmacokinetic model was used to simulate the steady-state pharmacokinetics of imatinib, which would be expected after ∼5 days of the 7-day dosing regimen. The population pharmacokinetic model was developed using a mixed-effects approach with a one-compartment model and was based on the plasma concentration profiles obtained for the single oral dose of 400 mg imatinib from the SAD portion of the study. The schedule of blood samples in the MAD study are detailed in figure 1. In the MAD study, the predicted imatinib steady-state exposure data obtained from the population pharmacokinetic model were compared to the observed imatinib steady-state systemic exposure for all doses of AV-101. The average concentration at steady state over one dosing interval (Cav) was used as an estimate to compare the inhaled AV-101 (10–90 mg twice daily) and oral imatinib (400 mg daily) dosing regimens.
Safety and tolerability
In the SAD study, vital signs (systolic and diastolic blood pressure, heart rate and respiratory rate) and peripheral oxygen saturation were evaluated pre-dose (at screening and day 0) and at multiple time points post-dose (at 20 min, 40 min, and 1, 2, 4 and 9 h and prior to pharmacokinetic sampling at 24, 48 and 72 h). Laboratory tests (haematology, biochemistry and urinalysis) were performed pre-dose (at screening and day 0), on days 1 and 2 and prior to discharge. Spirometry assessments (per American Thoracic Society guidelines) were performed at screening, prior to dosing, following the 20-min, 1-h and 4-h pharmacokinetic blood samples, and at day 4. The schedule of assessments for the MAD study are detailed in figure 1.
Statistical analyses
Descriptive statistics were used to summarise demographics and safety parameters. For each part of the study, natural log-transformed parameters for imatinib were assessed statistically for dose proportionality; parameters were considered dose proportional if the 90% confidence interval for the slope coefficient included 1. Log-transformed parameters for AV-101 (all doses) were compared with oral imatinib using ANOVA, with dose level as a fixed factor, using the Tukey–Kramer method of adjustment for multiple comparisons and a two-sided significance level of 5%.
Results
Participant demographics and disposition
A total of 82 participants were included: 48 in the SAD study and 34 in the MAD study. Two participants withdrew prematurely from the study; one participant in the SAD 30-mg cohort withdrew consent and one participant in the MAD 90-mg twice daily cohort withdrew due to an adverse event of vomiting (the subject's last dose was in the evening on day 1 and they withdrew from the study after a 24-h observation on day 3). Demographic characteristics from the SAD and MAD studies were generally well balanced (table 1, supplementary table S1).
Demographic characteristics
SAD pharmacokinetics
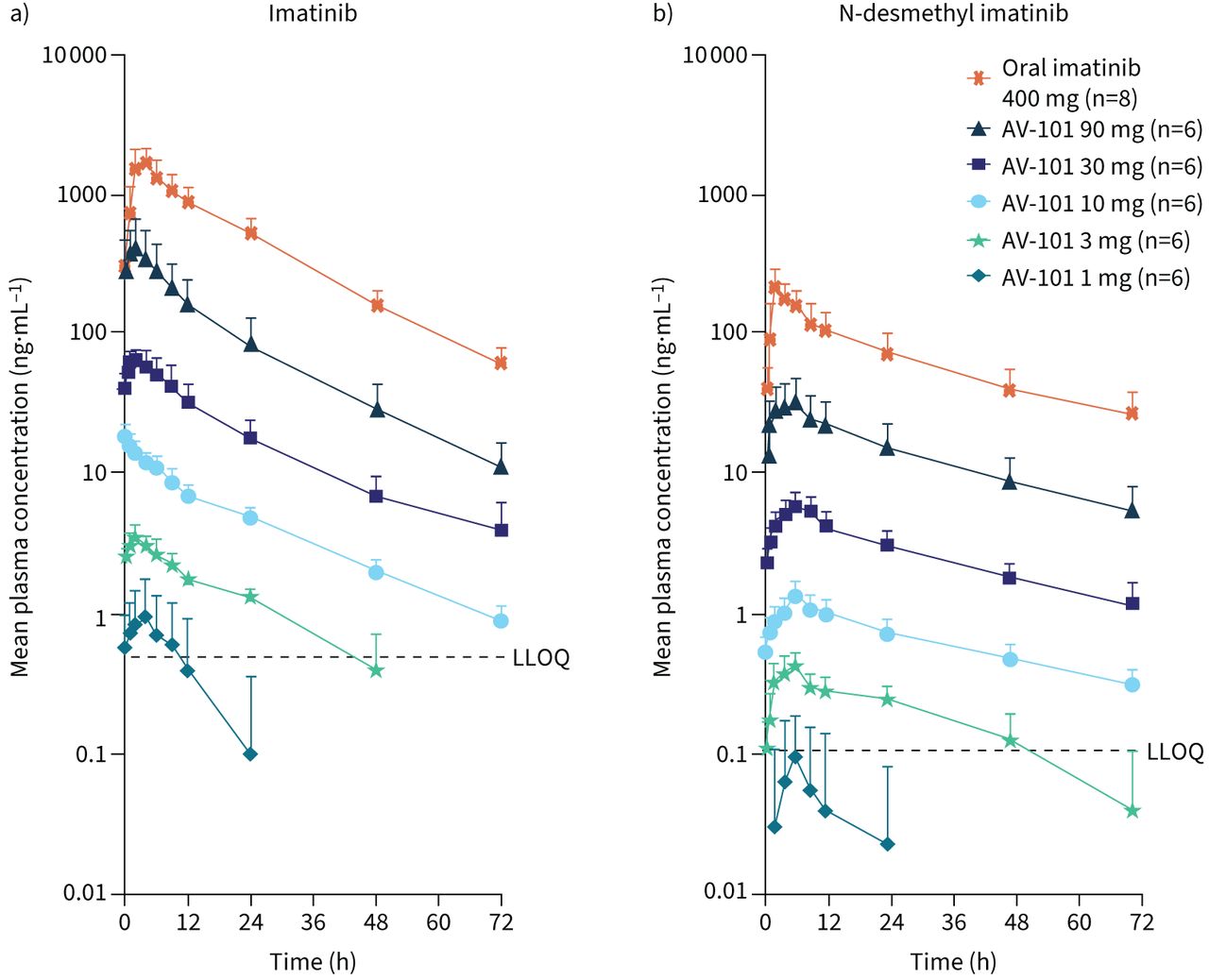
Following SADs of AV-101, the plasma concentrations of imatinib and N-desmethyl imatinib increased in a dose-dependent but greater than dose-proportional manner (figure 2, table 2). A 90-fold increase in imatinib exposure was observed across AV-101 dose levels, with statistical analysis confirming a greater than proportional increase in plasma imatinib exposure following AV-101 administration, as the 90% CI was >1 for both Cmax and AUC0-t (area under the concentration–time curve from dose administration at time 0 to tlast, where tlast is the time of last measurable observed concentration). At the 1-mg dose of AV-101, plasma concentrations of imatinib and N-desmethyl imatinib were below or near the assay's lower limit of quantification.

Concentration–time profiles for a) imatinib and b) N-desmethyl imatinib following single ascending dose of inhaled AV-101 or oral imatinib 400 mg. Data are presented as mean+sd over 72 h following administration on day 1. LLOQ: lower limit of quantification.
Pharmacokinetic parameters following single ascending dose of inhaled AV-101 or oral imatinib 400 mg
Plasma Cmax, AUC0-t and AUC0-∞ of imatinib and N-methyl imatinib following all AV-101 doses were consistently lower than those following a single dose of oral imatinib 400 mg, indicating significantly lower systemic exposure with AV-101 (p<0.001 for all doses). The plasma Cmax for imatinib was generally reached within 2–3 h across AV-101 dose levels, in contrast to 4 h following a single dose of oral imatinib 400 mg. The plasma Cmax for N-desmethyl imatinib was observed at 6 h for inhaled AV-101 and 2 h for oral imatinib. The plasma half-life for both imatinib and N-desmethyl imatinib was consistent across AV-101 dosing cohorts from 3 to 90 mg. The time of maximum observed concentration (tmax) for imatinib ranged from 2 to 3 h across AV-101 doses, with high variability except for the 10-mg dose, which resulted in a tmax of 0.18 (range 0.08–1.10) h. The tmax for N-desmethyl imatinib was ∼6 h for all AV-101 doses.
MAD pharmacokinetics
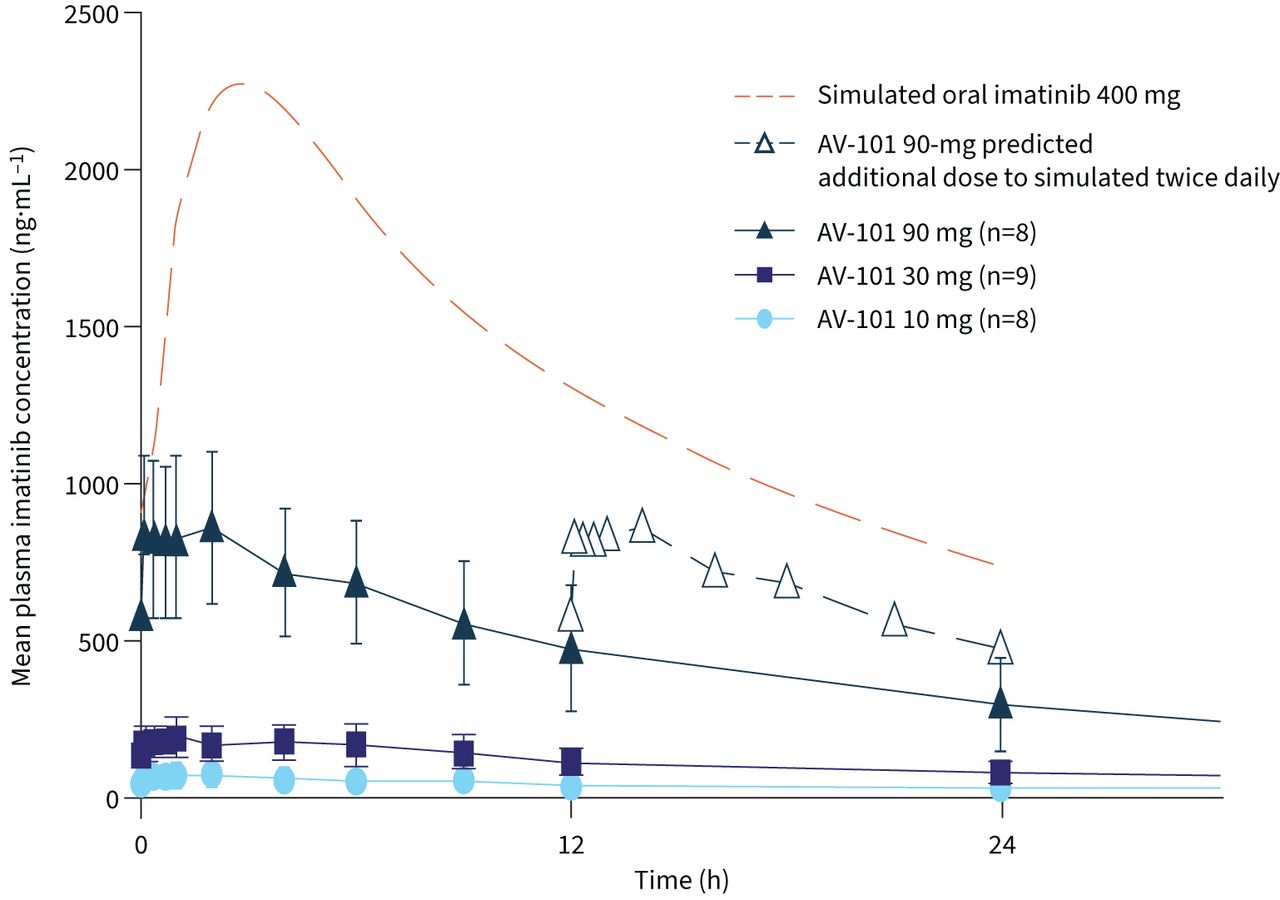
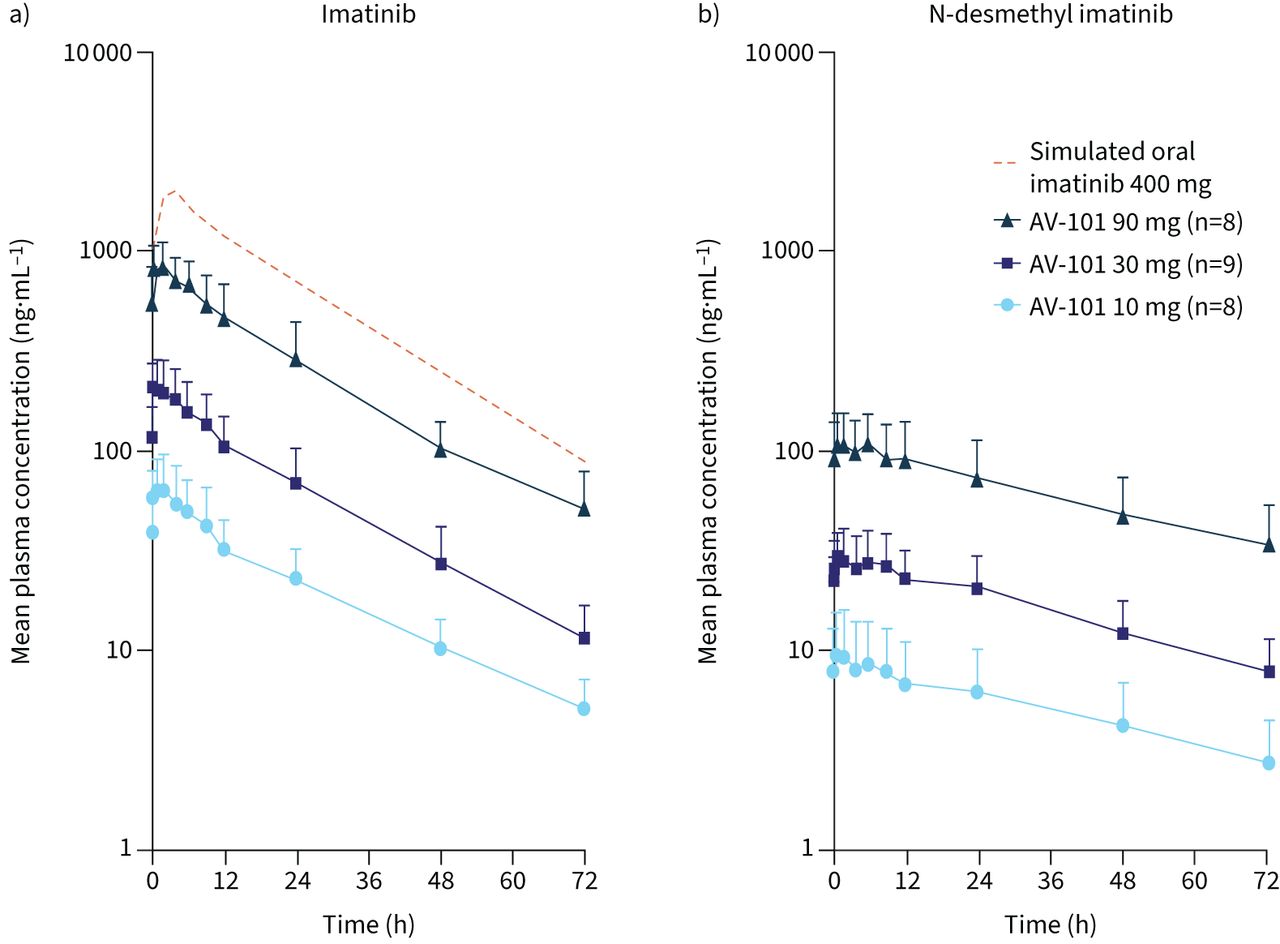
Systemic exposure of imatinib and N-desmethyl imatinib was higher following multiple versus single AV-101 doses, suggesting modest accumulation. Plasma concentrations of imatinib and N-desmethyl imatinib increased in a dose-proportional manner following multiple AV-101 doses (figure 3, table 3). While Cmax and AUCτ (area under the curve for the 0–12-h dosing interval at steady state) increased by 12.9-fold and 13.5-fold for imatinib, the corresponding increase was 11.7-fold and 12.0-fold for N-desmethyl imatinib, respectively, which was equivalent to a nine-fold increase from 10 mg to 90 mg AV-101. Statistical analysis confirmed the dose-proportional increase in plasma imatinib exposure, as the 90% CI contained or was close to 1 for both Cmax and AUCτ. Imatinib trough plasma concentration (Ctrough) following AV-101 administration reached a plateau by day 4. Overall, all AV-101 doses showed a lower steady-state systemic exposure (Cmax and AUCτ) compared with that modelled for oral imatinib at day 7 (p=0.0002); despite twice daily dosing, steady-state plasma concentrations for AV-101 90 mg remained below the simulated steady-state concentrations for oral imatinib 400 mg (figure 4).

Concentration–time profiles for a) imatinib and b) N-desmethyl imatinib following multiple ascending doses (day 7) of inhaled AV-101 or simulated oral imatinib 400 mg at steady state. Data are presented as mean+sd over 72 h following administration on day 7.
Pharmacokinetic parameters following multiple ascending doses (day 7) of inhaled AV-101 or simulated oral imatinib 400 mg at steady state
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Plasma concentrations of AV-101 (twice daily) and simulated oral imatinib 400 mg at steady state (day 7). Data are presented as mean±sd over 72 h following administration on day 7.
Safety and tolerability
In the SAD study, 10 TEAEs were experienced by nine (19%) participants, including eight who received AV-101 and one who received placebo; no participant who received oral imatinib 400 mg reported a TEAE. All TEAEs were grade 1 (mild); events following AV-101 included headache (n=3), dizziness (n=2), cough, diarrhoea, dyspnoea and nausea (n=1 each). No participant discontinued due to a TEAE.
In the MAD study, 32 TEAEs were experienced by 10 (29%) participants, including nine who received AV-101 and one who received placebo (table 4). All TEAEs were grade 1 or 2; all grade 2 TEAEs occurred in the 90-mg cohort and resolved by the end of the study. The most common TEAEs were cough and headache, occurring primarily in the 90-mg cohort. In this cohort, participants were required to complete nine inhalations at each dosing time point to achieve the 90-mg dose of AV-101; coughing typically began after three or four inhalations and resolved within a few minutes after the inhalations were complete. Only one participant experienced grade 2 coughing. One participant in the 90-mg cohort discontinued dosing on day 1 due to vomiting and withdrew from the study on day 3.
Multiple ascending dose study summary of treatment-emergent adverse events (TEAEs)
There were no clinically significant changes in spirometry, suggesting that coughing during AV-101 inhalations did not negatively impact lung function at 20 min or up to 4 h after dosing. In addition, there were no clinically significant changes in physical examination, vital signs, electrocardiogram or haematology, clinical chemistry and urinalysis values. Transient alterations in mean neutrophil counts within the normal range were observed in some dosing cohorts, but returned to baseline levels after 3–4 days.
Discussion
This study showed that systemic exposure to imatinib and its metabolite N-desmethyl imatinib was greatly reduced when imatinib was delivered as an inhaled dry powder (AV-101) using locally targeted doses of 1–90 mg versus oral administration of 400 mg in healthy adult participants, and that AV-101 was generally well tolerated. Following a single dose of AV-101, plasma concentrations of imatinib and N-desmethyl imatinib increased in a dose-dependent but greater than dose-proportional manner; following multiple doses of AV-101, plasma concentrations increased in a dose-proportional manner. Moderate plasma accumulation of imatinib and N-desmethyl imatinib was observed with repeated AV-101 doses; however, systemic exposure following AV-101 remained significantly lower than the observed single-dose and simulated multiple-dose, steady-state exposure of oral imatinib 400 mg.
As expected from the lower systemic exposure of imatinib, AV-101 was generally well tolerated with both single and repeated doses from 1 to 90 mg. In phase 2 and phase 3 studies with oral imatinib 400 mg, key systemic adverse events included gastrointestinal toxicities and oedema [24, 25]. In the present study, two participants (AV-101 90-mg twice daily cohort) experienced grade 1 nausea; no participant experienced oedema. While myelosuppression was observed in prior oral imatinib studies [24, 25], only transient increases and decreases in mean neutrophil counts (within normal range) were observed following AV-101. 6-month inhaled toxicology data in nonhuman primates also suggested no safety concerns following AV-101 (data on file). The more favourable safety/tolerability profile following AV-101 versus oral imatinib is not unexpected given the reduction in systemic imatinib exposure [29].
The most common TEAEs with AV-101 (MAD study) included cough and headache, primarily at the 90-mg twice daily dosage; this may reflect the nine inhalations per dose required for this cohort. Inhalation of drugs can cause sensitisation or direct irritant effects, including cough [30]. However, coughing events were typically mild and did not affect lung function, as spirometry assessments (forced expiratory volume in 1 s and forced vital capacity) performed 20 min to 4 h after dosing were normal. Multiple inhalations may also have led to the adverse event of vomiting and, consequently, the discontinuation of treatment by one participant in the AV-101 90-mg MAD cohort. To establish a practical dosing regimen and to mitigate potential coughing and headaches, future AV-101 studies (IMPAHCT; clinicaltrials.gov identifier NCT05036135) will decrease the amount of inhaled dry powder relative to the 90-mg dose by ≥60% and use only two versus nine inhalations per dose.
The determination of an appropriate dose that can be administered in a convenient and practical manner, while maintaining efficacious lung concentrations throughout a dosing period, is challenging. At the reference dose for oral imatinib (400 mg), efficacy and plasma levels are well established, but there are no corresponding lung concentration estimates over time. Furthermore, animal models do not translate well in terms of dose or disease pathology in humans, although aerosolised imatinib has exhibited positive responses in such models [31, 32]. Consequently, we relied on animal studies to generate lung and plasma time-course data and employed pharmacokinetic modelling to guide dosing estimates. In this regard, intratracheal instillation studies of imatinib in Sprague Dawley rats demonstrated high lung-to-plasma exposure ratios relative to orally administered imatinib (unpublished data). Additional pharmacokinetic parameters from rat and nonhuman primate data, as well as data from this SAD/MAD study, enabled construction of a semi-physiological, compartmental model based on work by Hendrickx et al. [33] to simulate lung exposure via inhalation as well as oral administration. These exercises provided further guidance on lung doses for the ongoing phase 2b/phase 3 study, and we expect that a 35-mg twice daily inhaled dose of AV-101 will achieve similar lung exposure to that of oral imatinib 400 mg. These estimates also account for extrapolation of lung-deposited doses to nominal capsule doses for use with the dry powder inhalers based on well-established in vitro techniques, such as inertial cascade impaction.
The observed plasma concentrations of inhaled AV-101 indicated rapid emergence of imatinib into the circulation, with appreciable plasma levels apparent 5 min post-dose. In contrast with 400 mg oral imatinib, plasma levels were not predicted to be observed for ≥20 min. These data provide confirmation that the source of imatinib is primarily via the lungs. Additionally, plasma levels appeared sustained for 4–6 h, although these levels may reflect an oral contribution (albeit small compared to a 400-mg oral dose) from inhaled drug that is deposited in the oropharynx and subsequently swallowed and absorbed. Nevertheless, the plasma concentration–time profiles of imatinib over the first 15–30 min are indicative of dissolution rate-limited absorption, otherwise a rapid initial decline in plasma levels would be expected since imatinib is considered a high-permeability drug [34]. These data are supported by intratracheal instillation studies in rats with suspension versus solution formulations of imatinib (data on file).
Numerous pre-clinical studies have shown kinase inhibitors to be effective in targeting pathways implicated in the pathogenesis of PAH (e.g. PDGF, fibroblast growth factor 2, vascular endothelial growth factor, epidermal growth factor, c-KIT and Src) [4, 16, 21], including imatinib, nilotinib, nintedanib, dasatinib and sorafenib [14, 15, 20, 21, 35, 36]. Despite promising pre-clinical results, the effectiveness of these other kinase inhibitors has largely not translated to the clinical PAH setting [37–41]. While oral imatinib demonstrated significant clinical benefit for PAH as add-on therapy in the IMPRES trial, it was associated with poor systemic tolerability that prohibited its further development. These data suggested that altering the delivery route of imatinib instead of the drug itself could lead to a more favourable benefit/risk profile.
In conclusion, this study showed that lower doses of imatinib via AV-101 dry powder inhalation resulted in significantly lower systemic exposure of imatinib in healthy adult participants compared to oral imatinib. Moreover, AV-101 was generally well tolerated with repeated dosing, supporting further development for treatment of PAH.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00433-2022.supplement
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: H. Gillies, R. Niven and B. Dake are employees of Aerovate Therapeutics, Inc. M.M. Chakinala received research grants/funding from Acceleron Pharma, Actelion, Eiger Biopharmaceuticals, Gossamer Bio, Medtronic and United Therapeutics Corporation; served as a consultant for Actelion, Altavant Sciences, Inc., Express Scripts Holding Company, Liquidia Technologies, Inc., PhaseBio Pharmaceuticals, United Therapeutics Corporation and WebMD LLC (Medscape). J.P. Feldman received honoraria from Acceleron Pharma, Altavant Sciences, Bayer, Gilead Sciences and United Therapeutics Corporation. M. Humbert received research grants/funding from Acceleron Pharma, Aerovate Therapeutics, Inc., Altavant Sciences, Inc., Bayer, Janssen Pharmaceuticals, Merck, Morphogen-IX Limited and United Therapeutics Corporation; and received honoraria from Acceleron Pharma, Actelion, Bayer, GlaxoSmithKline, Merck and United Therapeutics Corporation. M. Kankam is an employee of Altasciences Kansas, Inc.; and received research grants/funding from Actelion, Acurx, Biogen, BioXcel, DynPort Vaccine Company, Grifols, Jazz Pharmaceuticals, Novo Nordisk, Novus, Pfizer, Urovant Sciences, ViroDefense, and the US Food and Drug Administration/National Institutes of Health. N.S. Hill, M.M. Hoeper and V.V. McLaughlin have no disclosures to declare.
Support statement: This analysis and the studies included were sponsored by Aerovate Therapeutics, Inc. Medical writing and editorial assistance were provided by Tamara K. Stevenson (Lumanity Scientific Inc., Yardley, PA, USA), and were financially supported by Aerovate Therapeutics, Inc. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received August 26, 2022.
- Accepted October 16, 2022.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










