





Abstract
Diffuse panniculitis is a rare manifestation of α1-ATD, albeit perhaps the most fulminant and life-threatening complication, associated usually with ZZ phenotype. Intravenous α1-AT treatment is lifesaving. https://bit.ly/3EDmCzT
To the Editor:
Diffuse panniculitis is an inflammatory condition of the subcutaneous fat associated with a multiplicity of aetiological factors and nosological conditions [1, 2]. Diffuse panniculitis commonly occurs spontaneously and presents with painful skin nodules occasionally evolving into skin-ulcerating lesions discharging oily, yellow exudate. Histology shows an inflammatory infiltrate with lobular, septal or combined distribution, depending on the subjacent entity and the timing of biopsy, consisting of neutrophils, lymphocytes, histiocytes or a combination thereof; moreover, foamy macrophages, multinuclear giant cells, granulomas, necrosis or vasculitis may be seen [3]. Lesions may heal spontaneously (albeit less commonly) or after appropriate treatment with atrophic scarring. Any part of the superficial body may be involved although upper and lower extremities are more commonly affected [3]. Occasionally, it presents as part of a systemic inflammatory syndrome involving several extra-skin tissues and organs, and associates with thrombosis, a life-threatening clinical scenario [2].
A 17-year-old patient, a current smoker with a body mass index of 33.2 kg·m−2, presented to the emergency department complaining of erythematous, painful, indurating skin nodular lesions and low-grade fever over the previous 2 weeks (figure 1a). His previous medical and family history were noncontributory. Lesions were located bilaterally in the axillary regions, at the right abdominal surface and in both glutei. Differential diagnosis included mostly erythema nodosum and autoimmune rheumatic disease. After dermatological evaluation, a diagnosis of diffuse panniculitis was proposed. From laboratory evaluation, abnormal values included high C-reactive protein (CRP) (65 mg·L−1), low folic acid (1 ng·mL−1), low vitamin B12 (138 pg·mL−1), low albumin (2.7 mg·dL−1), low total serum proteins (4.8 mg·dL−1) and high ferritin levels (447 ng·mL−1). Computed tomography (CT) of the thorax and abdomen disclosed only diffuse oedema of the subcutaneous fat in the affected areas. Empirical antimicrobial treatment was started initially with a β-lactam, then changed to combined meropenem and linezolid upon deterioration. Due to low vitamin and albumin levels, enteric malabsorption was suspected, but endoscopy of the upper and lower gastrointestinal tract and capsule endoscopy for the small intestine did not detect abnormalities. Surgical biopsy of subcutaneous fat showed septal panniculitis with many histiocytes (figure 1b–d). Serology for autoimmunity, testing for HIV, hepatitis B virus and hepatitis C virus, and Quantiferon test were all negative. While lesions located in the glutei and the left axillary region became ulcerated, leaking oily, yellow exudates (figure 1a), the right abdominal wall lesion subsided spontaneously. New painful lesions appeared in concomitance with excessive subcutaneous oedema on the arms, scrotum and thighs. The lesions in the right arm and thighs ulcerated. Exudate cultures for common and specific pathogens including fungi and mycobacteria proved sterile. The patient continued to be febrile and his general clinical condition deteriorated. Due to excessive oedema of the arms and high D-dimer values, CT pulmonary angiography was performed, excluding pulmonary embolism. In the next few days and while lesions on the right upper arm appeared to subside, an insidious subcutaneous emphysema developed.
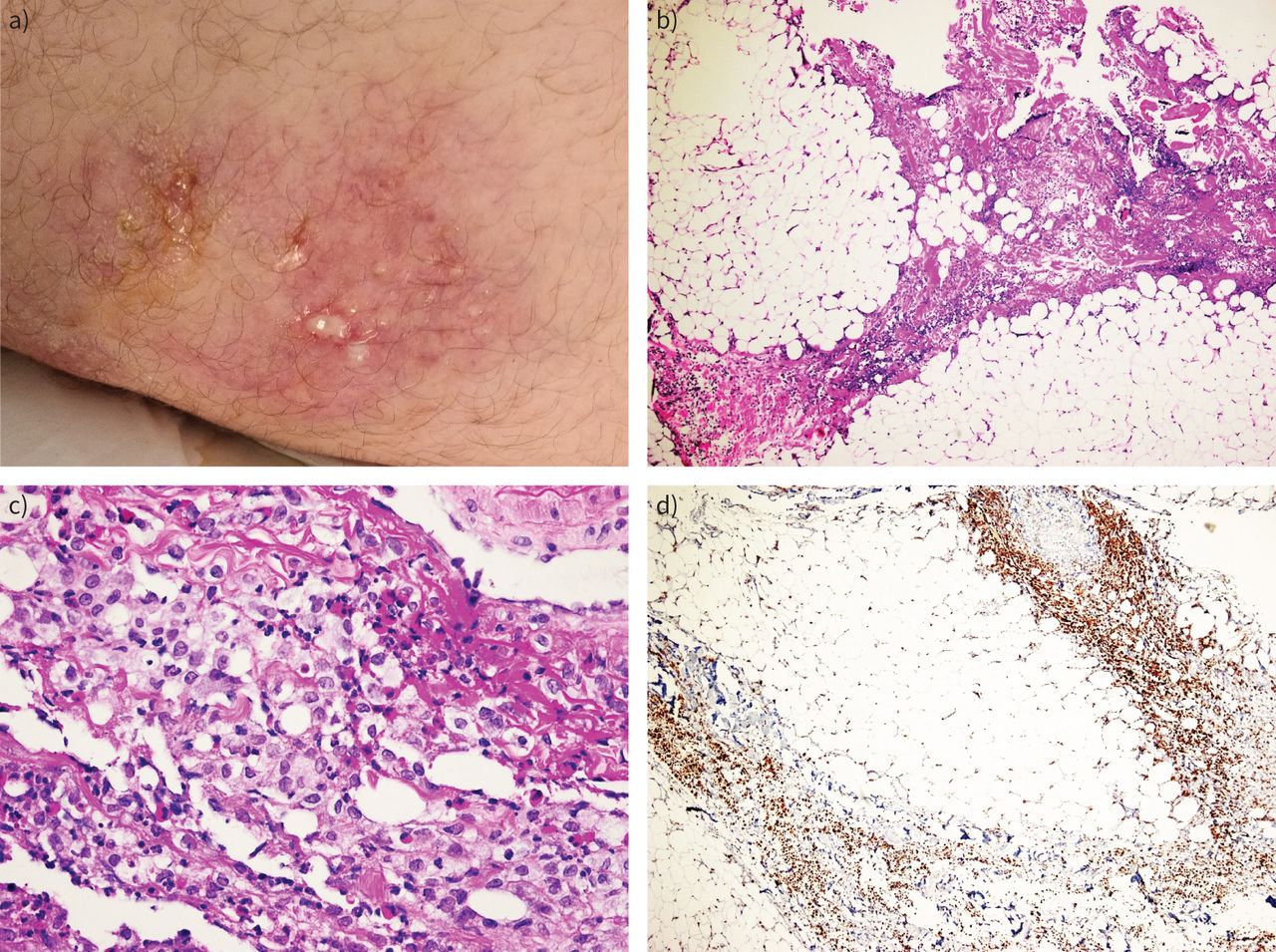
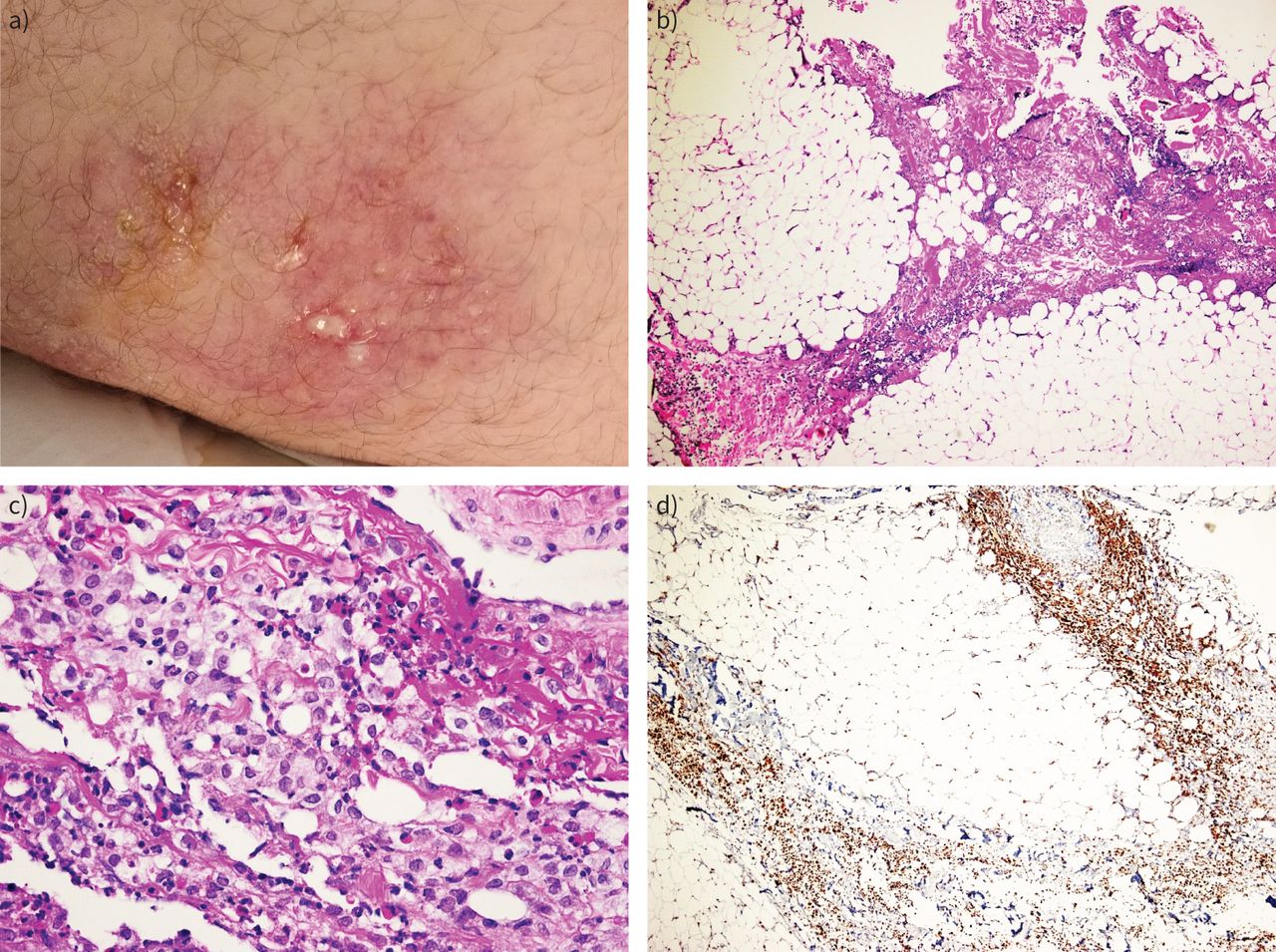{kind=link}
a) Erythematous, painful, indurating skin nodular lesions, partly ulcerated and leaking oily, yellow exudate. b) Panniculitis with inflammation mainly centring on fibrous septa (septal type) (haematoxylin and eosin (H&E) stain, 40× magnification). c) Histiocytic predominance in the inflammatory infiltrate (H&E stain, 40× magnification). d) Immunostaining for CD68 (PGM-1 clone) confirming histiocytic predominance (40× magnification).
In the course of diagnostic work-up, serum α1-antitrypsin (α1-AT) levels were found to be extremely low (0.3 g·L−1, normal values 0.9–2.0 g·L−1) and the diagnosis of α1-antitrypsin deficiency (α1-ATD)-associated diffuse panniculitis as part of a systemic inflammatory syndrome was established; isoelectric focusing and genotyping confirmed the ZZ genotype/phenotype. Pulmonary function testing was normal except a diffusing capacity of the lung for carbon monoxide of 68% predicted (systemic inflammatory syndrome with pleural effusions and obesity). Dapsone not being available for use, doxycycline was initiated, a course of plasmapheresis was performed and sequentially, intravenous augmentation infusion with α1-AT was started at a dosage of 100 mg·kg−1 after authorisation was obtained. The day after the initiation of the augmentation therapy, the patient presented remarkable improvement of his general condition. Rapid remission of the inflammatory lesions and normalisation of the laboratory tests followed. The patient was discharged, smoking cessation was recommended and the next administration of augmentation treatment was scheduled. On the day of readmission (the 10th day), a mild reactivation of the disease was evident, rapidly responding to the next i.v. α1-AT administration.
Diffuse panniculitis associated with α1-ATD is an extremely rare, underdiagnosed systemic manifestation, this is potentially severe or even lethal, associated usually with ZZ genotype [4, 5]. α1-AT is the most abundant serum and tissue circulating antiprotease, produced mainly by liver hepatocytes [6]. α1-AT acts as a protease inhibitor preferentially targeting excess human neutrophil elastase and, by protecting lungs connective tissue, prevents early emphysema development. α1-ATD is one of the most common genetic conditions and the Z variant in the homozygous state accounts for the 1–2% of all pulmonary emphysemas. Low or absent plasma levels and/or dysfunctional α1-AT molecules, including mutant Z molecules in the form of polymers, increase the risk of developing early pulmonary emphysema, liver disease and, rarely, other systemic manifestations, including diffuse panniculitis and systemic vasculitis [7]. Cigarette smoking is considered the major additional risk factor for emphysema development [8]. Warter et al. [9], in 1972, first described the association between diffuse panniculitis and α1-ATD. Since then, >100 patients have been described, mostly associated with the ZZ phenotype [4].
α1-AT is an effective inhibitor of several serine proteinases in addition to neutrophil elastase (its main target), such as cathepsin G, trypsin, chymotrypsin, plasminogen activator and serine proteinase-3 [6]. In addition, α1-AT is a very potent systemic anti-inflammatory molecule able to regulate neutrophilic chemotaxis, activation and degranulation, and affecting immune response, autoimmunity and apoptosis through its interactions with interleukin 8, leukotriene B4 and tumour necrosis factor α [10]. Severe deficiency alleles, such as PiZ, PiSSiiyama, PiMMalton and PiKKings, present low serum α1-AT levels not by reducing synthesis in the liver hepatocytes but by its excessive degradation in the endoplasmic reticulum in a great proportion and by the intracellular formation of polymers of the mutant protein [7]. The accumulation of the above, because of their toxicity (gain of function), relates to neonatal hepatitis syndrome, early-life cirrhosis and hepatocellular carcinoma. Milder deficiency alleles such as the PiS, PiI and PiQueen's form polymers but at a slower rate [11]. Circulating polymers of the mutant protein not only lose any antiprotease and anti-inflammatory function but acquire a new and potent proinflammatory action at sensitive sites of the body (lung, liver, subcutis and vessels) to induce, sustain and increase inflammation, and provoke tissue damage [12]. This combined mechanism, loss of plasma antiprotease potential due to the serum α1-AT levels (loss of function) and increase of protease burden due to Z polymers’ action on neutrophilic local inflammation (gain of function) are considered the pathogenetic mechanism of tissue damage in diffuse panniculitis [13]. This putative mechanism is further confirmed by the prompt and excellent response that augmentation therapy with i.v. α1-AT provides in almost all patients; offering of a fresh pool of wild, highly anti-inflammatory molecules reduces local inflammation and restores tissue damage [4, 14]. In our patient, immediately after the confirmation of the diagnosis of diffuse panniculitis related to the α1-ATD ZZ phenotype, doxycycline was initiated mainly for its anti-inflammatory and immune-regulatory actions, plasmapheresis in an attempt to eliminate polymers of the Z mutant protein from blood and tissues, and augmentation therapy in order to offer targeted anti-inflammatory action.
To conclude, we describe a rare manifestation of α1-ATD, albeit perhaps the most fulminant and life-threatening complication of α1-ATD in adults with the ZZ and, much more rarely, with the SZ and MZ phenotype. Biopsy of the lesions, serum α1-AT levels with CRP and phenotyping/genotyping are indispensable to confirm the diagnosis. In the setting of a high CRP level associated with the inflammatory syndrome in diffuse panniculitis, sometimes the α1-AT levels may increase to higher levels, further emphasising the need for genotyping or phenotyping to make the diagnosis of α1-ATD. The recognised treatment options include dapsone, tetracyclines, intravenous α1-AT, plasmapheresis (low case numbers) and liver transplant (low case numbers) [4, 5]. So far, augmentation therapy is life-saving treatment [4]. In perspective, new drugs like fazirsiran (an RNA-interfering molecule that acts by degrading α1-AT and Z-α1-AT mRNA, thereby significantly reducing Z-α1-AT protein synthesis in hepatocytes and, therefore, leakage of Z polymers in plasma and tissues) may find a place in the treatment of α1-ATD-related diffuse panniculitis [15] in combination with contemporaneous treatment with intravenous α1-AT.
Acknowledgements
Maria Sfika, Vasiliki Apollonatou (both National and Kapodistrian University of Athens, Athens, Greece), Lykourgos Kolilekas (Chest Diseases Hospital of Athens Sotiria, Athens, Greece), Emmanouil Korakas, Dimitrios Lygkos and Prof. Konstantinos Triantafyllou (all three National and Kapodistrian University of Athens, Athens, Greece) for patient care and assistance.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: S.A. Papiris made major contributions to the concept and design of the study, and to the acquisition, analysis and interpretation of data, and wrote the manuscript; A. Parmaxidis made major contributions to the management of the patient, and to the acquisition, analysis and interpretation of data, and wrote part of the manuscript; S. Theotokoglu and Z. Tsakiri made major contributions to the documentation of the diagnosis, to the analysis and interpretation of data, and wrote part of the manuscript; M. Veith performed the genetic analysis for the patient, and made major contributions to the interpretation of data and revised critically this work for very important intellectual content; A. Parmaxidis, V. Pappa and M. Kallieri made major contributions to the management of the patient and interpretation of data, and revised critically this work for very important intellectual content; J-F. Mornex made major contributions to the interpretation of data and revised this work critically for very important intellectual content; A.C. Katoulis had major contribution to the documentation of the diagnosis, analysis and interpretation of data and revised critically this work for very important intellectual content; D. Haritos made major contributions to the management of the patient, and the acquisition, analysis and interpretation of data, and critically revised this work for very important intellectual content; I.G. Panayiotides made major contributions to the documentation of the diagnosis, analysis and interpretation of data, drafted part of the manuscript, and critically revised this work for very important intellectual content; E.D. Manali made major contributions to the concept and design of the study, to the acquisition, analysis and interpretation of data, drafted part of the manuscript, had access to all data, supervised the accuracy and integrity of any part of the work and revised critically this work for very important intellectual content. All authors read and approved of the final version of the submitted manuscript.
Conflict of interest: S.A. Papiris declares payment or honoraria from DEMO SA, in the 36 months prior to manuscript submission. J-F. Mornex declares consulting fees from CSL Behring, Grifols, LFB and Takeda; and payment or honoraria and support for attending meetings from CSL Behring, Grifols and LFB, all in the 36 months prior to manuscript submission; and that they are a member of the scientific board of ADAAT. E.D. Manali declares support for attending meetings from CSL Behring in the 36 months prior to manuscript submission. All other authors declare no competing interests.
- Received October 17, 2022.
- Accepted November 17, 2022.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org










