





Abstract
Background Interleukin-11 (IL-11) is linked to the pathogenesis of idiopathic pulmonary fibrosis (IPF), since IL-11 induces myofibroblast differentiation and stimulates their excessive collagen deposition in the lung. In IPF there is disrupted alveolar structural architecture, yet the effect of IL-11 on the dysregulated alveolar repair remains to be elucidated.
Methods We hypothesised that epithelial-fibroblast communication associated with lung repair is disrupted by IL-11. Thus, we studied whether IL-11 affects the repair responses of alveolar lung epithelium using mouse lung organoids and precision-cut lung slices (PCLS). Additionally, we assessed the anatomical distribution of IL-11 and IL-11 receptor (IL-11R) in human control and IPF lungs using immunohistochemistry.
Results IL-11 protein was observed in airway epithelium, macrophages and in IPF lungs, also in areas of alveolar type 2 (AT2) cell hyperplasia. IL-11R staining was predominantly present in smooth muscle and macrophages. In mouse organoid co-cultures of epithelial cells with lung fibroblasts, IL-11 decreased organoid number and reduced the fraction of Prosurfactant Protein C-expressing organoids, indicating dysfunctional regeneration initiated by epithelial progenitors. In mouse PCLS exposed to IL-11, ciliated cell markers were increased. The response of primary human fibroblasts to IL-11 on gene expression level was minimal, though bulk RNA-sequencing revealed IL-11 modulated various processes which are associated with IPF, including unfolded protein response, glycolysis and Notch signalling.
Conclusions IL-11 disrupts alveolar epithelial regeneration by inhibiting progenitor activation and suppressing the formation of mature alveolar epithelial cells. Evidence for a contribution of dysregulated fibroblast–epithelial communication to this process is limited.
Abstract
Interleukin-11 negatively impacts lung epithelial regeneration by inhibiting progenitor cell activation and suppressing alveolar differentiation https://bit.ly/3ZRDpIR
Introduction
Idiopathic pulmonary fibrosis (IPF) is believed to be induced by repetitive micro-injuries to the alveolar epithelium leading to defective epithelial-fibroblast communication and chronic activation of extracellular matrix (ECM)-producing myofibroblasts [1]. Dysfunctional alveolar type 2 cells (AT2 cells) are believed to play a fundamental role in the initiation of fibrosis [2, 3]. In IPF, the regenerative capacity of AT2 cells is impaired [3], with loss of both AT1 and AT2 cells observed in IPF tissue. Additionally, AT2 cells in IPF were reported to show abnormalities such as senescence and cellular stress [1, 4].
Interleukin-11 (IL-11) is a member of the IL-6 family and is associated with cancer and fibrosis in numerous organs [5, 6]. Interestingly, IL-11 protein levels are hardly detectable in healthy individuals [7–9], suggesting IL-11 is not essential for homeostasis. Although fibroblasts and epithelial cells have been proposed as sources of IL-11 in the lung [9, 10], it is not yet established which cell types primarily produce IL-11 and express IL-11 receptor (IL-11R) in the human lung. Recent mouse and human studies suggest that IL-11 plays a fundamental role in IPF pathogenesis. IL-11 can induce fibroblast to myofibroblast transition and stimulate collagen secretion by myofibroblasts, and the fibrotic response in a bleomycin mouse model was diminished by the administration of an IL-11 antibody [9].
Since IL-11 was found to influence IPF pathogenesis by acting on fibroblasts [9], we hypothesised it also negatively impacts normal epithelial lung repair. We first explored the protein expression pattern of IL-11 and its receptor in human lung tissue and assessed differences between control and IPF tissue. We then studied the influence of IL-11 on epithelial progenitor cell function using a mouse organoid model. Finally, we examined whether IL-11 induced dysfunctional fibroblast–epithelial communication, thereby affecting progenitor cell behaviour.
Methods
Full details of materials and methods are described in the online supplementary material.
Animal handling
Specific pathogen-free wild-type C57BL/6J mice (>8 weeks) were used in this study. All animal experiments were performed according to the national guidelines and upon approval of the experimental procedures by the local Animal Care and Use committee of the University of Groningen.
Immunohistochemistry
The immunohistochemical staining performed in this manuscript were conducted as part of the HOLLAND (HistopathOLogy of Lung Aging aNd COPD) project, and were based on existing protocols [11].
Briefly, sections from paraffin-embedded human lung tissue of control and IPF donors (table 1) were incubated with rabbit anti-IL-11 (LSBio, Seattle, WA, USA) or rabbit anti-IL-11R (Abcam, Cambridge, UK), diluted 1:100 or 1:400, respectively, and subsequently incubated with peroxidase-conjugated donkey anti-rabbit (Jackson Immunoresearch, Ely, UK) diluted 1:500. Sections were developed with NovaRED (Vector Laboratories, Burlingame, CA, USA), followed by haematoxylin counterstain. Slides were scanned with a Hamamatsu NanoZoomer 2.0HT digital slide scanner (Hamamatsu Photonic K.K., Hamamatsu City, Japan) at magnification 40×. Per staining, the mean intensity and percentage of positive stained area was quantified using Fiji/ImageJ 2.1.0 [12].
Patient characteristics of human lung tissue samples stained for interleukin (IL)-11 and IL-11 receptor
Mouse epithelial cell isolation and organoid culture
Epithelial (CD31−/CD45−/EpCam+) cells were isolated as previously described [13], and the organoid assay was based on published protocols [13]. In this manuscript, two types of organoids were used; mouse organoids (primary mouse EpCam+ cells combined with CCL206 fibroblasts) and mixed organoids (primary mouse EpCam+ cells co-cultured with primary human fibroblasts; table 2). Briefly, 10 000 fibroblasts were mixed with 10 000 freshly isolated mouse lung EpCam+ cells and seeded into transwell inserts (Thermo Fischer Scientific, Waltham, MA, USA) in 100 μL growth factor-reduced Matrigel (Fisher Scientific, Landsmeer, The Netherlands) diluted 1:1 with DMEM:Ham's F12 (1:1) containing 10% FBS, 2 mM L-glutamine and antibiotics. Organoids were exposed to recombinant human IL-11 (rhIL-11, R&D Systems, Minneapolis, MN, USA, #218-IL) for 14 days. Organoids were fixed and stained according to published protocols [13].
Patient characteristics of primary human fibroblasts used for organoid culture, gene expression studies and RNA sequencing
Precision-cut lung slices
Mouse lungs were filled with 1.5% low-melting point agarose (Gerbu Biotechnik GmbH, Wieblingen, Germany) solution. Subsequently, lung lobes were cut into precision-cut lung slices (PCLS) of 250 µm in thickness. Slices were incubated with 100 ng·mL−1 rhIL-11 for 48 h, after which they were stored at −80°C until further use.
Gene expression analyses
300 000 primary fibroblasts of control and IPF donors (table 2) of passage 5 until 7 were exposed to rhIL-11 for 24 h, after which their RNA was extracted using TRIzol (Invitrogen under Thermo Fisher Scientific). The Maxwell simplyRNA tissue kit (Promega, Madison, WI, USA) was used to isolate RNA from mouse PCLS. Equal amounts of mRNA were reverse transcribed (Promega) and real time quantitative PCR (qPCR) was performed.
RNA sequencing
300 000 primary human lung fibroblasts of control donors (table 2) of passage 5 until 7 were exposed to 100 ng·mL−1 rhIL-11 for 24 h. RNA was isolated using TRIzol reagent. An Illumina NovaSeq 6000 sequencer was used for bulk RNA-seq data analysis by GenomeScan (the Netherlands). Differentially expressed genes, pathway enrichment analysis and volcano plots were generated in R. The complete dataset is available via https://doi.org/10.6084/m9.figshare.19419707.v1.
Data analyses
Data are presented with mean±sd unless described otherwise. A two-tailed t-test, a one-way ANOVA, a two-way ANOVA, a Wilcoxon test or a Kruskall–Wallis test was used where appropriate. Differences were considered statistically significant when p<0.05.
Results
IL-11 and IL-11R expression in human lung tissue
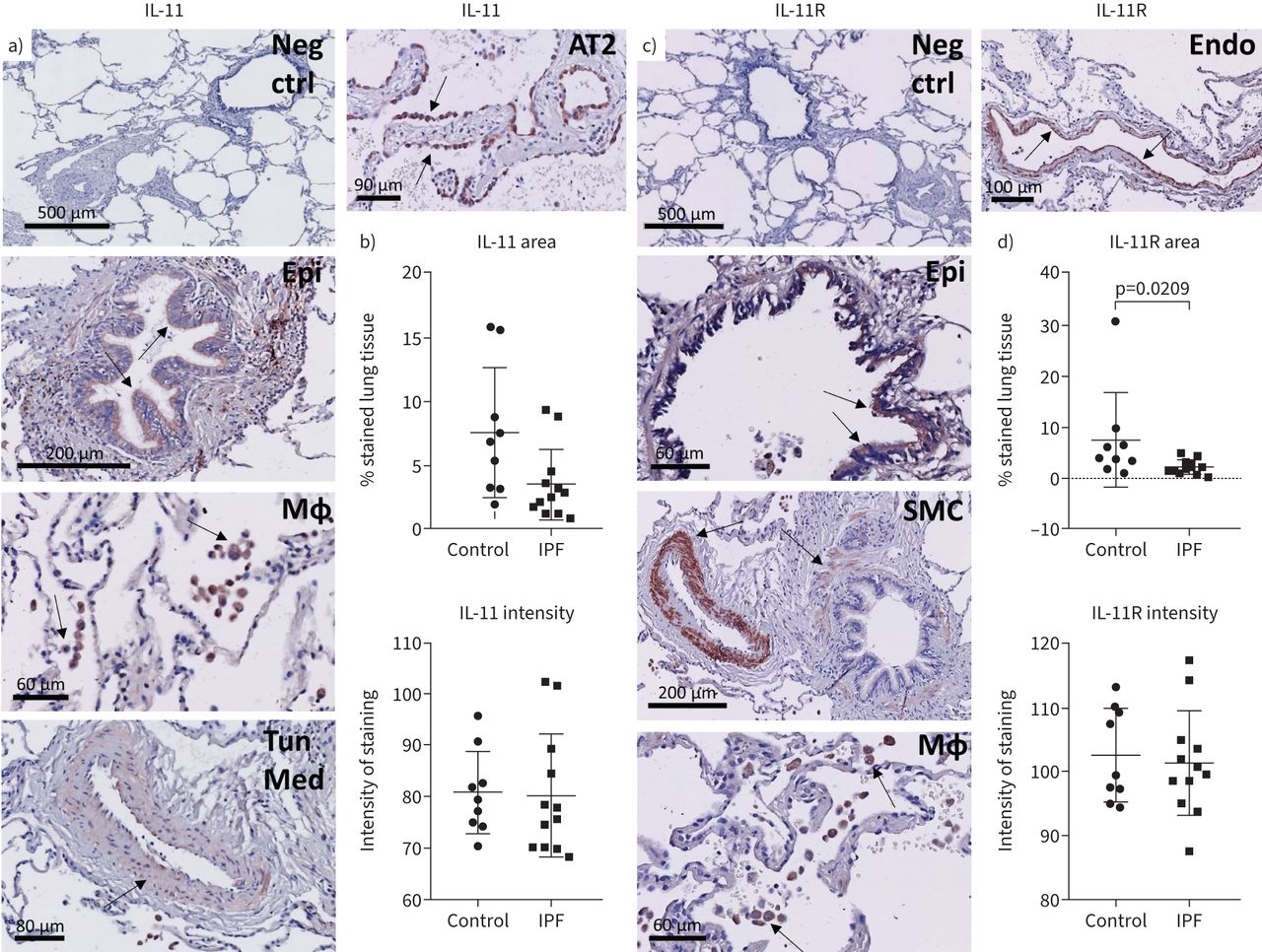
The cell type-specific expression of IL-11 and IL-11R in the human lung remains incompletely understood, though publicly available RNA-sequencing datasets suggest fibroblasts, myofibroblasts, innate lymphoid cells, endothelial cells and various epithelial cells express IL-11, whereas IL-11R is expressed by a variety of cells, including epithelial cell types, fibroblasts, smooth muscle cells, dendritic cells and endothelial cells (supplementary figure S1). These publically available data indicate no notable differences between controls and IPF donors with the exception of aberrant basaloid cells which are unique to IPF donors and appear to express IL11. Other cell types such as the innate lymphoid cell type A are more abundant in IL11 expression when obtained from healthy donors. We further investigated the expression pattern on protein level by performing immunohistochemical staining for IL-11 and IL-11R in human lung tissue. First, secondary antibody specificity was confirmed (figure 1a, c). IL-11 protein expression was observed in the airway epithelium, a subset of macrophages and to a lesser extent in the tunica media of blood vessels and in the smooth muscle around airways (figure 1a). In IPF tissue, there was also some IL-11 staining in AT2 cells in areas of AT2 cell hyperplasia (figure 1a). Quantitative analysis of IL-11 tissue positive area and average intensity of staining (figure 1b) indicated that there were no overall differences between control and IPF tissue (p=0.2401 and p=0.8256 respectively). IL-11R staining was primarily visible in macrophages and in smooth muscle surrounding vessels and airways, and in IPF tissue, also in randomly organised smooth muscle. IL-11R was also present to a lesser extent in airway epithelium and the endothelium (figure 1c). Both IL-11 and IL-11R staining were not observed in fibroblast foci in IPF tissue. The IL-11R staining area was significantly decreased in IPF lung tissue compared with control tissue (figure 1d) (p=0.0209), whereas the intensity of IL-11R staining did not differ between groups (p=0.7016). For the staining quantification of both IL-11 and IL-11R, the variability within each group was substantial. Interestingly, numerous control and IPF tissue sections were almost completely negative for IL-11 and/or IL-11R staining, whereas others did show positivity. We noticed heterogeneity not only within the groups, but even within the same tissue, as structures were mostly only partially positive for IL-11 and IL-11R (supplementary figures S2–S3). For example, generally only a subset of macrophages and AT2 cells were positive within one tissue section, and protein expression could be found in the epithelium of one airway, whereas other airways present in the tissue were negative. We also assessed whether IL-11 and IL-11R were present in the exact same area of the same tissue by comparing serial sections (supplementary figure S4). Exact overlap was sometimes observed, though it was more common that IL-11 and IL-11R were present in non-identical but adjacent structures.

Expression pattern of interleukin (IL)-11 and IL-11 receptor (IL-11R) in the human lung. Human lung tissue sections of control and idiopathic pulmonary fibrosis (IPF) donors were immunohistochemically stained for IL-11 and IL-11R, developed with NovaRED (red) and counterstained with haematoxylin (blue). Arrows indicate positive cells or structures. Negative control images (Neg ctrl) are included for both IL-11 and IL-11R (a, c). a) IL-11 staining was present in airway epithelium (Epi), macrophages (MΦ) and the tunica media of blood vessels (Tun Med) (example images are from control donors). In IPF, IL-11 staining was also present in areas of alveolar type 2 cell hyperplasia (AT2) (a). b) Analysis of IL-11 staining area and intensity in control (n=9) and IPF (n=12) tissue, unpaired t-test on log transformed data. c) IL-11R expression was observed in airway epithelium (Epi), smooth muscle (SMC), macrophages (MΦ) and the endothelium (Endo). All example images of IL-11R are from the control group. d) Analysis of IL-11R staining area and intensity in control (n=9) and IPF (n=12) tissue, unpaired t-test on log transformed data.
IL-11 negatively impacts progenitor activation and alveolar differentiation
Since little is known about the influence of IL-11 on alveolar repair, we used a mouse organoid model to study the regenerative capacity of epithelial progenitor cells in response to IL-11 (figure 2a). IL-11 concentrations of 0–10 ng·mL−1 were reported in nasal secretions of children with respiratory infections, whereas in vitro cultures of fibroblasts and epithelial cell lines mostly produced up to 10 ng·mL−1 IL-11, though sometimes concentrations of around 100 ng·mL−1 were found [7, 14]. Therefore, we used concentrations of 1–100 ng·mL−1 IL-11 in the organoid system. Overall, the number of organoids formed on day 14 was significantly decreased in the presence of rhIL-11 (p=0.0045). Post hoc analyses showed that 10 ng·mL−1 and 100 ng·mL−1 rhIL-11 both induced a significant reduction in the number of organoids formed (p=0.0303 and p=0.0224 respectively) (figure 2b). The organoid number data demonstrated remarkable variability. We hypothesised this may be caused by sex differences; however the dataset was mostly comprised of epithelial cells originating from female mice (n=8 out of n=10). Therefore, we expanded our experiments to include more data from male mice, and then stratified the number results for mouse sex from which the epithelial cells were isolated (figure 2c). Although rhIL-11 significantly reduced the number of organoids formed (p=0.0017), the response to rhIL-11 was not dependent on the sex of the donor mouse. In addition, the median organoid diameter on day 14 was not altered in the presence of rhIL-11 (p=0.3819) (figure 2d).

Recombinant human interleukin (IL)-11 suppresses epithelial progenitor cell activation in mouse organoids. a) Representative brightfield images of mouse organoids exposed to rhIL-11. Scale bar=200 µm. b) Normalised mouse organoid number on day 14 after continuous treatment with a dose curve of rhIL-11 (n=10, 2 male, 8 female; one-way ANOVA with Sidak's post hoc test on log transformed data). c) Normalised number of mouse organoids in response to rhIL-11, separated by mouse sex (n=11 male, n=8 female; two-way ANOVA with Sidak's post hoc test on log transformed data). d) Organoid diameter on day 14 in response to rhIL-11; median is shown (n=10, Kruskall–Wallis test with Dunn's post hoc test).
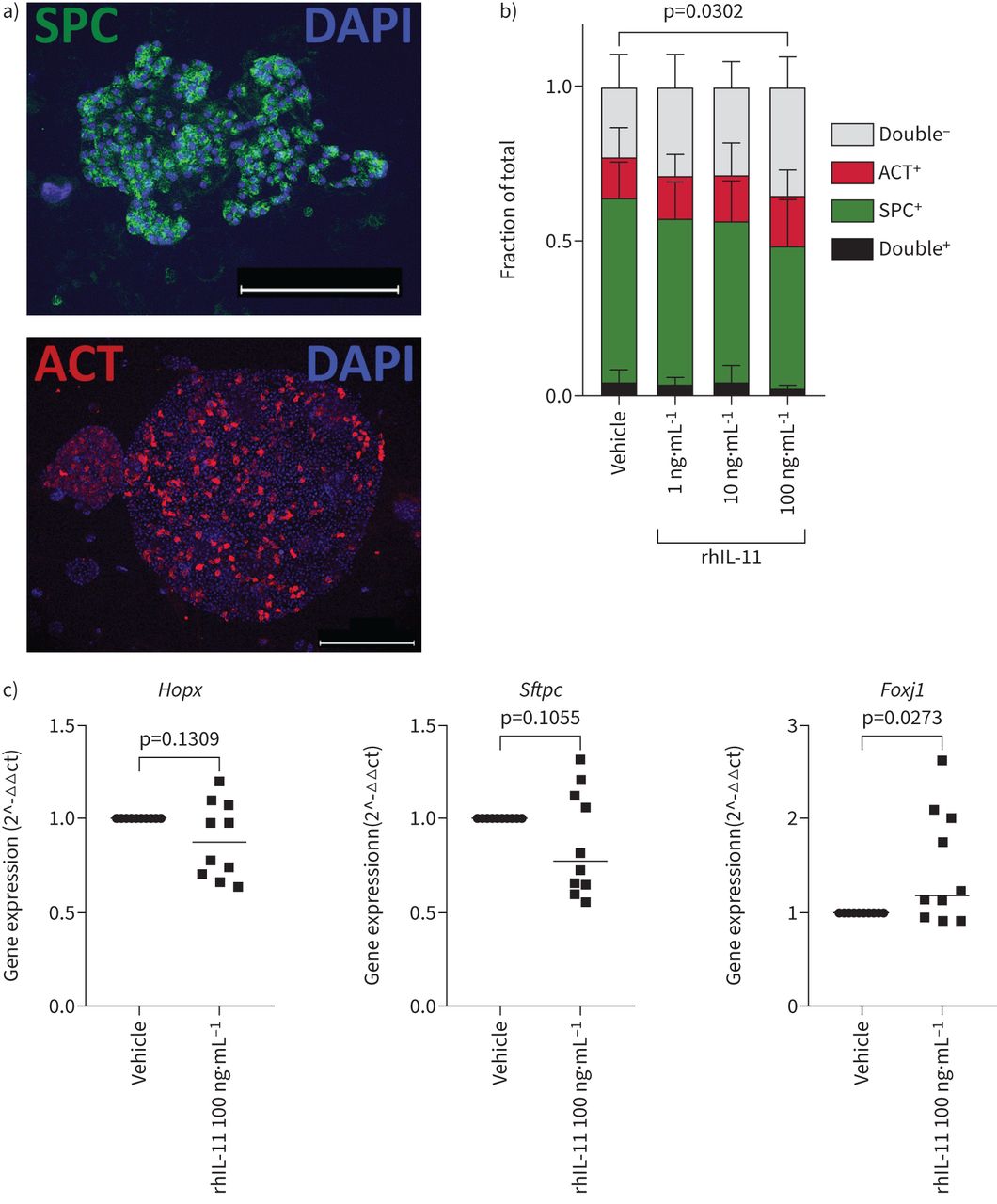
To assess the effect of IL-11 on epithelial differentiation, immunofluorescence was performed for differentiation markers Prosurfactant Protein C (pro-SPC) and Acetylated α Tubulin (ACT) (figure 3a). Quantification of these markers revealed that rhIL-11 significantly influenced their expression (p=0.0302), where 100 ng·mL−1 rhIL-11 induced a reduction of the pro-SPC-expressing organoid fraction (p=0.0089) and concurrently an increase in the double-negative fraction (pro-SPC–ACT–organoids) (p=0.0126) (figure 3b).

Interleukin (IL)-11 dysregulates epithelial cell differentiation. a) Representative fluorescent images of a Prosurfactant Protein C (pro-SPC)+ (scale bar=50 µm) and Acetylated α Tubulin (ACT)+ (scale bar=100 µm) organoid. b) Quantification of the fraction of pro-SPC-expressing and ACT-expressing mouse organoids in response to recombinant human (rh)IL-11 (n=10, two-way ANOVA with Sidak's post hoc test). c) Precision-cut lung slices of wild-type mice were exposed to 100 ng·mL−1 rhIL-11 for 48 h, after which RNA was isolated from the whole slice and gene expression studies were performed. Gene expression of alveolar epithelial cell markers Hopx and Sftpc, and of ciliated cell marker Foxj1, median is shown (n=10, paired samples Wilcoxon test on delta ct values).
We then assessed the response of mouse organoids to rmIL-11 (supplementary figure S5A), as the species of recombinant IL-11 was previously suggested to be of importance for studying its effects [15]. Overall, rmIL-11 also affected the amount of organoids formed on day 14 (p=0.0010), where 1 ng·mL−1 significantly increased the number of organoids (p=0.0098) and 100 ng·mL−1 significantly reduced the number of organoids formed (p=0.0321) (supplementary figure S5B). Organoid size was not affected by rmIL-11 exposure (supplementary figure S5C). Next, we compared the expression of Il11ra1 in the two cell types present in the organoid system, the EpCam+ cells and CCL206 fibroblasts, by culturing control mouse organoids for 72 h after which cell types were resorted into EpCam+ cells and CCL206 fibroblasts and bulk RNA-sequenced separately. The expression level of Il11ra1 was considerably higher in the CCL206 cells than in the EpCam+ cells (supplementary figure S6A). We also checked the expression of Il11, which was relatively low in both cell types. We continued by investigating the response of CCL206 fibroblasts to IL-11, by exposing them to both rmIL-11 and rhIL-11 for 30 min, after which cells were lysed and subjected to Western blot. An increase in phosphorylated STAT3 and ERK1/2 was observed in CCL206 fibroblasts after rmIL-11 and rhIL-11 treatment, indicating activation of downstream signalling pathways JAK/STAT3 and MEK/ERK (supplementary figure S6B–D). The CCL206 response to rhIL-11 was more pronounced compared to rmIL-11, and the effects of rhIL-11 on mouse organoid formation were also stronger and more consistent. Therefore, we continued our studies with rhIL-11.
After observing indications of progenitor cell dysfunction induced by IL-11 in the organoid model, we assessed whether IL-11 could also affect the epithelium in mouse PCLS. After incubating the slices with rhIL-11 for 48 h, we assessed the gene expression of epithelial cell markers. The expression of alveolar cell markers (Hopx and Sftpc) was not significantly affected by the presence of rhIL-11 (p=0.1309 and p=0.1055 respectively) (figure 3c), though similar to the organoid number data described in figure 2b, the expression of these markers showed large variability, where the PCLS obtained from some mice responded more strongly to IL-11 than others. Whereas the expression of Sgb1a1 and Muc5ac was unaffected by rhIL-11, Foxj1 expression was significantly increased (p=0.0273) (supplementary figure S7A, figure 3c). We also assessed expression of Il11 and Il11ra1 and genes associated with fibrosis (supplementary figure S7B–C), and found that most genes were not altered in response to rhIL-11, though Tgfb1 expression was downregulated (p=0.0039).
Next, we wondered if the IL-11-induced aberrant progenitor response that we observed in mouse organoids would also occur using a combination of primary mouse EpCam+ cells and primary human fibroblasts (figure 4a), and whether the disease state of the fibroblasts would affect the response to IL-11. Importantly, the co-culture of primary mouse EpCam+ cells and primary human fibroblasts was previously shown to yield organoids comparable in number, size and differentiation to those formed by primary mouse EpCam+ cells combined with CCL206 fibroblasts [13]. Overall, rhIL-11 exposure induced a significant decrease in the number of organoids (p=0.0001) formed by primary mouse EpCam+ cells combined with primary human fibroblasts (figure 4b). This was not dependent on whether the fibroblasts used in the cultures were isolated from control or IPF tissue (p=0.3055). There was no effect on organoid diameter in cultures established with either control or IPF fibroblasts (figure 4c–d), similar to our previous results.

The mouse epithelial progenitor cell response is disturbed by interleukin (IL)-11 in mouse/human organoid co-cultures. Primary mouse Epcam+ cells mixed with primary human lung fibroblasts (either control or idiopathic pulmonary fibrosis (IPF)) were seeded in Matrigel, cultured for 14 days and continuously exposed to rhIL-11; resulting organoid formation was studied. a) Representative brightfield images of organoids formed by mouse EpCam+ cells with control primary human fibroblasts, in response to recombinant human (rh)IL-11. Scale bar=250 µm. b) Normalised number of mouse/human organoids in response to 14-day treatment with rhIL-11 (n=5 control fibroblasts, n=5 idiopathic pulmonary fibrosis (IPF) fibroblasts; two-way ANOVA with Sidak's post hoc test on log transformed data). c) Diameter of organoids formed by mouse Epcam+ cells with control fibroblasts after rhIL-11 treatment, median is shown (n=5, Kruskal–Wallis test with Dunn's post hoc test). d) The effect of rhIL-11 on the diameter of organoids formed by mouse Epcam+ cells with IPF fibroblasts, median is shown (n=5, Kruskal–Wallis test with Dunn's post hoc test).
The production of known epithelial progenitor supportive factors by fibroblasts is not affected by IL-11
Since the organoid is a co-culture system, the observed effects of IL-11 in this system (figures 2 and 3) could be determined by an epithelial or by a fibroblast response. As the staining pattern of IL-11R (figure 1) and the gene expression levels of Il11ra1 in control mouse organoids (supplementary figure S6A) suggest that alveolar epithelial cells express IL-11R to a limited extent, we hypothesised that the organoid effects could be indirect and regulated by fibroblasts. Thus, we exposed primary human fibroblasts (either control or IPF) to rhIL-11 for 24 h after which gene expression studies were performed. The expression of progenitor supporting factors and fibrosis-marker gene Fibronectin 1 (FN1) in response to rhIL-11 (supplementary figure S8A-B) was variable, with no significant effects on any measured genes. The expression of TGFB1, IL11 and IL11RA was also not influenced by IL-11 treatment of either control or IPF fibroblasts (supplementary figure S8B–C).
IL-11 affects processes in fibroblasts including metabolism, cellular stress and cross-talk mechanisms
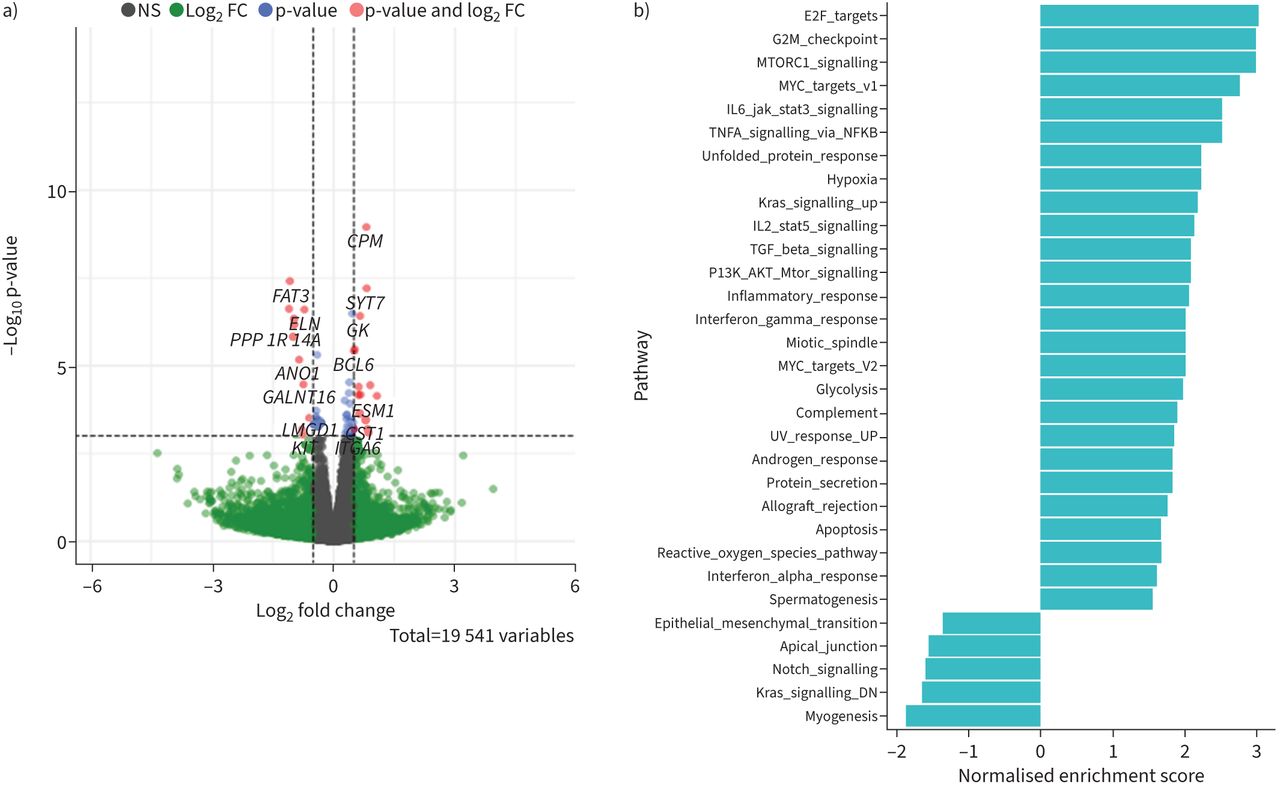
We then performed bulk RNA-sequencing to get a broader overview of the influence of IL-11 on human fibroblasts. The volcano plot (figure 5a) shows there were relatively few genes that were differentially expressed in response to 24-h exposure to rhIL-11 (24 genes with adjusted p-value (padj) <0.05). Significantly upregulated genes include Hexokinase 2 (HK2) and Lactate Dehydrogenase A (LDHA), which are involved with glycolysis, and Suppressor Of Cytokine Signaling 3 (SOCS3), which is a target gene of STAT3 and acts as a negative feedback mechanism of JAK/STAT signalling [16]. Gene Set Enrichment Analysis (GSEA) revealed IL-11 significantly upregulated pathways associated with proliferation (E2 factor (E2F) targets, G2M checkpoint, MYC targets and mitotic spindle) (figure 5b), which was previously shown to be increased in fibroblasts by IL-11 [17], and downstream mechanisms of IL-11 signalling; JAK/STAT3, MEK/ERK (KRAS) and PI3 K/Akt/mTOR [16], indicating IL-11 induces expected responses in primary fibroblasts. Other processes that were affected by IL-11 include TGFβ signalling, inflammation (TNFα signalling via NF-κB, inflammatory response, IFNγ response, complement and IFNα response), cellular stress (unfolded protein response and reactive oxygen species pathway), metabolism (glycolysis) and cross-talk pathways (Notch signalling).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Interleukin (IL)-11 alters several mechanisms in primary human fibroblasts which can influence fibrosis and cellular cross-talk. Primary human fibroblasts of control donors (n=5) were treated with 100 ng·mL−1 rhIL-11 for 24 h and subsequently subjected to bulk RNA-sequencing analysis. a) Volcano plot showing significantly altered genes in control fibroblasts treated with IL-11 (log2fold change (FC) cut-off=0.5 and adjusted p-value (padj) cut-off=0.05). b) Gene set enrichment analysis (GSEA) showing upregulated pathways (positive enrichment score) and downregulated pathways (negative enrichment score) induced by IL-11.
Discussion
In this study we describe for the first time the distribution of IL-11 and IL-11R in human lung tissues on protein level, and show that IL-11 impairs progenitor cell activation and suppresses alveolar differentiation, suggestive of dysregulated alveolar regeneration. We also explored mechanisms by which fibroblast–epithelial cross-talk may be disturbed by IL-11, but we found little evidence for this in gene expression studies.
Immunohistochemical staining of IL-11 and IL-11R was present in various cell types, and we note considerable intra- and interindividual variability in the expression of both proteins. The observations made here regarding the IL-11- and IL-11R-expressing cells and structures are not fully in line with previous literature [9, 15, 18–25], which might be due to previous studies mainly reporting gene expression data, often in cell cultures or mouse organs, whereas here protein expression in human lung tissue was assessed. Interestingly, IL-11 expression was also present in macrophages, which are suggested to be of importance in the pathogenesis of IPF [26–28]. We also noted IL-11 expression in areas of AT2 cell hyperplasia (figure 1a), which is of particular interest since epithelial cells are thought to produce IL-11 [9, 10] and epithelial cell dysregulation relates to the initiation of fibrosis [10]. Whereas IL-11 mRNA overexpression has been reported in IPF lungs [9], here we found no differences in IL-11 protein between control and IPF tissues. The percentage of tissue area positive for IL-11R was significantly decreased in IPF compared to control. The reason for this is not known since the regulation of IL-11R is largely unexplored [29], though the disorganisation of the lung architecture in IPF compared to healthy lung tissue may affect the overall expression of IL-11 and IL-11R as well. Furthermore, the donors of the control and IPF tissues differed in smoking status, pack-years and years since smoking cessation (table 1), which could also affect IL-11(R) expression. Though IL-11R expression was not observed in fibroblast foci, fibroblasts activate downstream signalling pathways in response to IL-11 [9], suggesting IL-11R expression is dynamically regulated and may differ between cellular subsets or activation states, highlighting the need for studies on IL-11R regulation.
IL-11 decreased the number of organoids formed, indicating disrupted progenitor cell activation. IL-11 also reduced the pro-SPC+ organoid fraction, which indicates AT2 cell dysfunction or disturbances in alveolar differentiation. It was previously shown that human embryonic stem cell-derived organoids that model Hermansky–Pudlak syndrome-associated interstitial pneumonia (HPSIP), which also recapitulates features of IPF, had an abnormal morphology which was restored in IL-11 knockout HPSIP organoids [10]. Moreover, IL-11 was recently reported to inhibit primary human alveolar cell proliferation, whereas primary mouse AT2 cells treated with IL-11 had an increased expression of KRT8 and stalled transdifferentiation to AT1 cells, supporting our hypothesis that IL-11 impairs alveolar repair [30]. Interestingly, we found that IL-11-exposed PCLS showed an upregulation of Foxj1 expression. Though this finding warrants more study, it may relate to bronchiolisation, a pathological process in IPF where alveolar spaces are covered with bronchiolar cell types [1], as microscopic honeycombing was previously found to be specifically associated with increased expression of cilium-associated genes in a subset of IPF patients [31]. Since IL-11 induced distinct epithelial differentiation responses in organoids compared to PCLS, IL-11 may affect epithelial cells differently during regeneration or repair (modelled by the organoid) than during homeostatic conditions (modelled by the PCLS system). Our data indicating that IL-11 affects organoid number but not size further suggests that IL-11 impacts on the progenitor cell decision to form an organoid but not on the subsequent proliferative expansion phase.
Rather, since lung epithelial progenitor cell behaviour is strongly influenced by the microenvironment [32], we hypothesised that IL-11 dysregulates fibroblast–epithelial cell cross-talk in relation to repair. This is supported by a recent publication which shows that IL-11 activates downstream signalling more strongly in fibroblasts than in colon epithelial organoids, and that IL-11+ fibroblasts differentially express epithelial supportive factors such as HGF and EREG [33]. Though we found little evidence for a disrupted progenitor support function of fibroblasts in our studies on the gene expression level, Notch signalling was downregulated in fibroblasts upon IL-11 exposure. Sustained activation of Notch is related to abnormal epithelial repair with alveolar cyst formation [33]. However, alveolar repair relies on dynamic Notch signalling, where Notch is initially active to stimulate progenitor cell activation after which it is inactivated to support differentiation [33]. Thus, dysregulated Notch signalling may have implications for abnormal re-epithelialisation. Pathways involved with inflammation, such as TNFα signalling via NF-κB and inflammatory response were upregulated in fibroblasts exposed to IL-11. IL-11 is known to have immunomodulatory effects [18, 34]. However, the role of inflammation in IPF is not completely clear [1]. IL-11 also upregulated HK2, LDHA and the glycolysis pathway, the reactive oxygen species pathway, the unfolded protein response and the TGFβ pathway, which have all been associated with IPF previously [35–38].
A limitation of our study is that IL-11 did not induce large effects on gene expression level, which was previously reported as well [9]. This prevented us from exploring whether fibroblast–epithelial communication mechanisms were distorted by IL-11 using RNA-seq. Proteomics would be a potential approach to give new insights to these questions. Furthermore, we describe immunohistochemical staining patterns of IL-11 and IL-11R, but cell types such as quiescent fibroblasts are difficult to recognise without specific markers, and could therefore not be characterised. Finally, the expression and response of IL-11 showed large variability in our studies. Since IPF is known to be more prevalent in men than in women [1], we hypothesised that sex differences could play a role in the response to IL-11. However, this was not supported by our organoid data, though we only investigated possible effects of sex differences in the EpCam+ cells and not in the fibroblasts. If IL-11 were to be used as a therapeutic target in the future, the heterogeneity of IL-11 should be addressed.
In summary, IL-11 disturbs alveolar organoid formation as a model of alveolar regeneration in vitro, which may relate to dysfunctional epithelial repair responses in IPF. Though we did not find evidence on gene expression level for a role of IL-11 in disrupting the epithelial progenitor support function of fibroblasts, we show that various other cell types also express IL-11 and IL-11R in the human lung, including epithelial cells, macrophages and smooth muscle cells, and their IL-11-driven responses related to fibrosis and dysregulated epithelial repair will be interesting to uncover.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00679-2022.SUPPLEMENT
Acknowledgements
We thank Sophie Bos (Department of Molecular Pharmacology, University of Groningen, Groningen, the Netherlands) for technical support. The stainings of interleukin (IL)-11 and IL-11 receptor on lung tissue performed in this manuscript were conducted as part of the HOLLAND (Histopathology of Lung Aging and COPD) project. The HOLLAND project was initiated and supervised by Corry-Anke Brandsma (University of Groningen, University Medical Center Groningen, Department of Pathology and Medical Biology, Groningen, the Netherlands), Wim Timens and Janette Burgess; technical support was provided by Marjan Reinders-Luinge (University of Groningen, University Medical Center Groningen, Department of Pathology and Medical Biology, Groningen, the Netherlands), Anja Bakker (University of Groningen, University Medical Center Groningen, Department of Pathology and Medical Biology, Groningen, the Netherlands) and Theo Borghuis; and image analyses pipelines were developed by Theo Borghuis, Maunick Lefin Koloko Ngassie (University of Groningen, University Medical Center Groningen, Department of Pathology and Medical Biology, Groningen, the Netherlands) and Niek Bekker (University of Groningen, University Medical Center Groningen, Department of Pathology and Medical Biology, Groningen, the Netherlands).
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: R.K. Kortekaas declares funding for the present manuscript from Boehringer Ingelheim (unrestricted research funds to their institute) and an internship contract with Boehringer Ingelheim in the 36 months prior to manuscript submission. K.E. Geillinger-Kästle is an employee of Boehringer Ingelheim. M. Webster is a former employee of Boehringer Ingelheim and a current employee of Newcells Biotech. W. Timens declares consulting fees from Merck Sharp & Dohme and Bristol Myers Squibb in the 36 months prior to manuscript submission; and that they are a board member of the Dutch Society of Pathology, and a member of the Council for Research and Innovation of the Federation of Medical Specialists. J.K. Burgess declares funding for the present manuscript from Boehringer Ingelheim (unrestricted research funds to their institute) and Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Aspasia-premie subsidienummer 015.013.010, paid to their institute). R. Gosens declares funding for the present manuscript from Boehringer Ingelheim (paid to their institute); as well as grants paid to their institution from Aquilo and Sanofi-Genzyme in the 36 months prior to manuscript submission. All other authors declare no competing interests.
Support statement: This research was supported by an unrestricted research grant from Boehringer Ingelheim to the University of Groningen.
- Received December 6, 2022.
- Accepted March 8, 2023.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










