



Clinical effectiveness of once-daily fluticasone furoate/umeclidinium/vilanterol in usual practice: the COPD INTREPID study design
- Sally Worsley1⇑,
- Neil Snowise2,3,
- David M.G. Halpin4,
- Dawn Midwinter5,
- Afisi S. Ismaila6,7,
- Elaine Irving1,
- Leah Sansbury6,
- Maggie Tabberer8,
- David Leather2 and
- Chris Compton2
- 1Medical Engagement & Value Evidence and Outcomes, GlaxoSmithKline plc., Stevenage, UK
- 2Global Respiratory Franchise, GlaxoSmithKline plc., Brentford, UK
- 3Faculty of Life Sciences and Medicine, King's College, London, UK
- 4Dept of Respiratory Medicine, Royal Devon and Exeter Hospital, Exeter, UK
- 5Clinical Statistics, GlaxoSmithKline plc., Stockley Park, UK
- 6Medical Engagement & Value Evidence and Outcomes, GlaxoSmithKline plc., Collegeville, PA, USA
- 7Dept of Health Research Methods, Evidence and Impact, McMaster University, Hamilton, ON, Canada
- 8Medical Engagement & Value Evidence and Outcomes, GlaxoSmithKline plc., Stockley Park, UK
- Sally Worsley, Medical Engagement & Value Evidence and Outcomes, GlaxoSmithKline plc., Gunnels Wood Rd, Stevenage, SG1 2NY, UK. E-mail: sally.d.worsley{at}gsk.com
Abstract
Effectiveness studies complement conventional randomised controlled trials by providing a holistic view of treatments in the setting of usual clinical practice. We present the protocol for the ongoing INTREPID (INvestigation of TRelegy Effectiveness: usual PractIce Design; ClinicalTrials.gov identifier: NCT03467425) study, a randomised, open-label, 24-week effectiveness study of once-daily fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI; Trelegy) delivered by the ELLIPTA inhaler versus non-ELLIPTA multiple-inhaler triple therapy in patients with chronic obstructive pulmonary disease (COPD) in usual practice settings. INTREPID was designed to provide evidence of FF/UMEC/VI effectiveness in patients with COPD managed in routine healthcare systems across multiple European countries. Between study initiation and end-of-study visits, patients will receive their medication and care as they would ordinarily receive it, from their usual healthcare provider at their usual healthcare centre. Study-specific intervention will be minimal. The primary end-point will be the proportion of COPD assessment test (CAT) responders, defined as a clinically meaningful improvement from baseline of ≥2 units, at week 24. The CAT was chosen as it provides health status information relevant to patients, physicians, health technology agencies and payers. Lung function (forced expiratory volume in 1 s) and critical inhaler errors will also be assessed in a subgroup of patients. The strengths and weaknesses of the protocol and some of the challenges associated with conducting this multicountry study, such as differences in healthcare systems and treatment practices across sites, will also be discussed.
Abstract
The INTREPID COPD study is investigating the real-world effectiveness of fluticasone furoate/umeclidinium/vilanterol delivered by ELLIPTA versus multiple-inhaler triple therapies in Europe. This article presents the protocol and discusses effectiveness studies in COPD. http://bit.ly/2ZzBAo3
The importance of randomised controlled trials and factors limiting their applicability to usual clinical practice
The outcomes of double-blind randomised controlled trials (RCTs) have driven the regulation of treatments made available to patients since the late 1940s [1]. However, it is now widely accepted that the stringent conditions under which these trials are conducted, such as highly selected patient populations, limiting comorbidities and concomitant medications of patients included, and the selection of end-points used, may make it challenging to extrapolate results directly into the context of usual practice [2]. This makes interpreting clinical trial results challenging for physicians when deciding upon the best treatment choice for individuals, and can potentially lead to reduced treatment benefits among patients.
Adherence to chronic obstructive pulmonary disease (COPD) therapy, inhaled or otherwise, is also higher in conventional RCTs (up to 90%) than in usual practice (10–40%) [3]. This is due to increased clinical supervision in RCTs compared with usual practice, the impact of the Hawthorne effect [4], which describes changes in patient behaviour due to an awareness of being studied, and subconscious selection biases in patient identification. Factors limiting the ability of RCTs to reflect usual practice and the ways in which effectiveness studies differ from RCTs are shown in table 1.
A comparison of the key features that differ between conventional randomised controlled trials (RCTs) and effectiveness trials
An introduction to effectiveness studies
Effectiveness studies provide data complementary to that derived from conventional RCTs by providing information on relative effectiveness, defined as the extent to which an intervention does more good than harm compared with alternative interventions when provided in usual practice [12]. Data from effectiveness studies provide valuable information to support health technology evaluations of new medicines and physician decision-making regarding what treatments to provide [13]. These studies can be categorised as interventional or noninterventional (observational).
Observational studies, including cohort, case–control and cross-sectional studies, are often used in the post-authorisation phase to provide insights into the real-world effectiveness, safety and tolerability of interventions over long periods of time in large patient populations [14]. The lack of randomisation and potential presence of selection bias in observational studies may, however, be considered a weakness of this study type and means that these studies have limited traction with regulators. Selection bias can complicate the establishment of clear cause-and-effect relationships due to underlying differences in patient characteristics, including differences in disease risk factors, indications for treatment and illness severity (low internal validity) [15]. The baseline characteristics and outcome data used in observational effectiveness studies are also limited to those available in the chosen database or cohort, which may not provide a complete picture of confounding factors that may influence data interpretation.
Issues of bias and confounding are partly overcome in randomised, interventional effectiveness trials (pragmatic trials), which are randomised trials undertaken in usual practice settings. Interventional effectiveness studies include the rigour of site training and data collection required by conventional RCTs, but balance this with minimal departure from usual practice. Randomisation increases the internal validity of these studies by reducing bias, while alignment of the population enrolled, end-points employed, comparators used and study visit schedules with standard-of-care guidelines facilitates the balancing of internal validity with generalisability [16]. A recent example of a randomised effectiveness study is the Salford Lung Study in COPD (SLS COPD), which investigated the safety and efficacy of once-daily fluticasone furoate/vilanterol (FF/VI) compared with continuing usual care across 74 general practice clinics in a single city in the United Kingdom (UK) [17, 18].
Rationale for the INTREPID study
The Global Initiative for Chronic Obstructive Lung Disease strategy report recommends triple therapy with an inhaled corticosteroid (ICS), a long-acting muscarinic antagonist (LAMA) and a long-acting β2-agonist (LABA) (ICS/LAMA/LABA triple therapy) for patients with symptomatic COPD at risk of exacerbations [19]. The improved ability of ICS/LAMA/LABA therapy to reduce exacerbation rates versus dual regimens (ICS/LABA or LAMA/LABA) has been demonstrated previously [20–25].
Until recently, such combinations were available only via multiple inhalers. Multiple-inhaler regimens are associated with reduced treatment adherence among patients [26] and a greater proportion of handling errors that, subsequently, can reduce treatment efficacy compared with single-inhaler therapies [27, 28]. The advent of single-inhaler triple therapy (SITT), for example with FF/umeclidinium (UMEC)/VI [29] or beclometasone/formoterol/glycopyrronium bromide [30], affords the opportunity to administer inhaled triple therapy in a simple regimen that may reduce inhaler errors among patients and thereby improve overall treatment benefits [27]. Patient satisfaction may also be improved with SITT, particularly among those who have difficulties with taking multiple medications or those who struggle with implementing correct inhaler technique [31].
The efficacy of FF/UMEC/VI has been thoroughly investigated in conventional RCTs. Of note, the recent, phase III FULFIL (lung FUnction and quality of LiFe assessment in COPD with closed trIpLe therapy) and IMPACT (InforMing the PAthway of COPD Treatment) trials demonstrated consistent and superior results for FF/UMEC/VI versus dual therapy with ICS/LABA or LAMA/LABA (budesonide/formoterol (FULFIL), FF/VI and UMEC/VI (IMPACT)) in patients with symptomatic COPD and a history of exacerbations [24, 25]. As conventional RCTs, however, FULFIL and IMPACT were unable to fully assess the effectiveness of FF/UMEC/VI in usual practice settings [32–34]. These studies also did not compare FF/UMEC/VI with any multiple-inhaler triple therapy (MITT) in conditions of low adherence [3] and high critical error rates, as is the case in usual practice. INTREPID (ClinicalTrials.gov identifier: NCT03467425) was designed to address this gap in our knowledge of FF/UMEC/VI and to allow the expansion of COPD effectiveness data from that obtained in SLS COPD to multiple sites in multiple European countries.
INTREPID: protocol of a randomised effectiveness study in usual practice
Study design
INTREPID is a randomised, open-label, phase IV effectiveness study designed to evaluate once-daily FF/UMEC/VI (100 μg/62.5 μg/25 μg) delivered by the ELLIPTA inhaler compared with any approved non-ELLIPTA MITT in patients with COPD in usual practice settings. INTREPID will be conducted at centres in Germany, the Netherlands, Spain, Sweden and the UK from April 2018 to December 2019 (planned finish). A key criterion in selecting countries was the ability to access patient data from electronic health records.
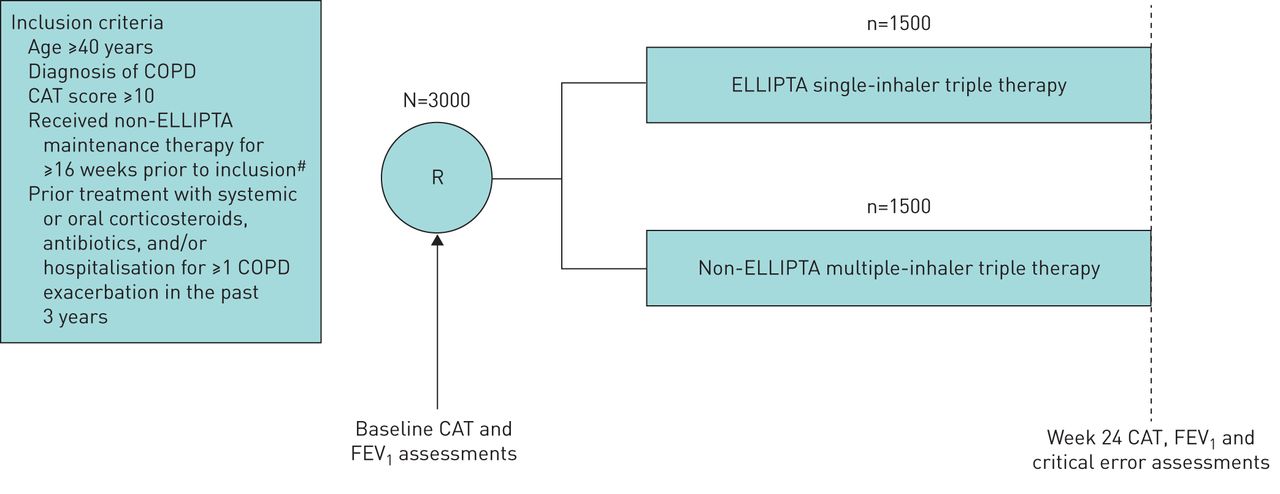
Patients meeting the inclusion criteria (table 2) will be enrolled into the study and randomised 1:1 to a prescription treatment (initiate FF/UMEC/VI or non-ELLIPTA MITT) for 24 weeks (figure 1). The inclusion and exclusion criteria aim to facilitate the inclusion of a broad patient population reflective of usual prescribing in accordance with the FF/UMEC/VI licence. To minimise the potential effects of study participation on the recommended usual care of patients with COPD, patients have only two mandated study visits: one at screening/randomisation and one at the study end (week 24 or early withdrawal). During the study, patients will see their treating physician according to usual care only (e.g. for repeat prescriptions, general medical care and exacerbations). Details of COPD and any asthma history, historical eosinophil levels, COPD medications and healthcare resource utilisation (HRU), including primary and secondary healthcare visits, will be recorded in an electronic case report form (eCRF) during any routine care or study visit. Further HRU data will be collected from national databases in the UK, the Netherlands and Sweden.
Key inclusion and exclusion criteria of the INTREPID study
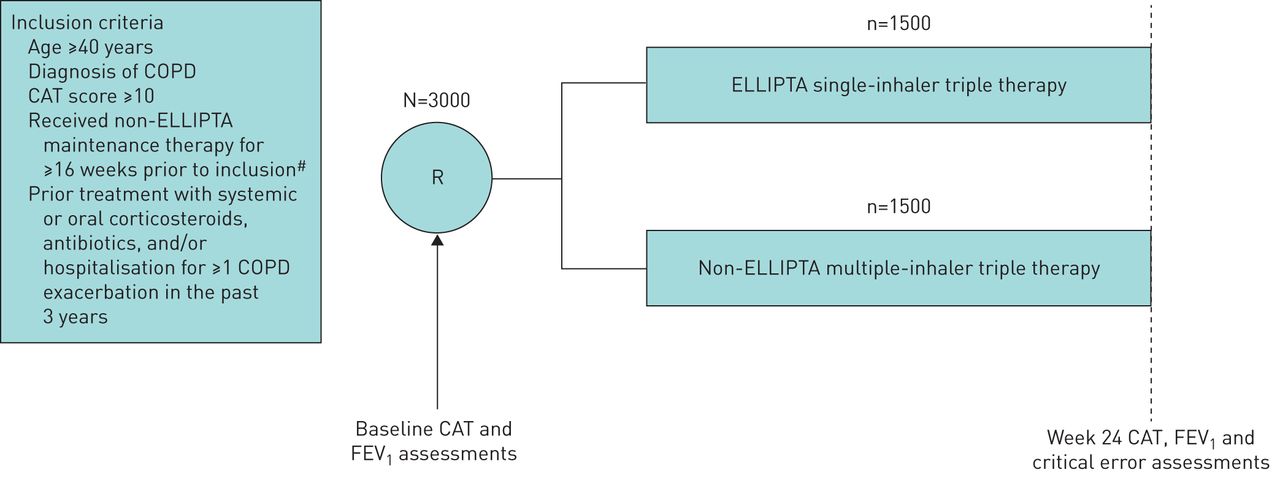
INTREPID study design. Only two study visits (baseline and week 24) will be required during the INTREPID study. FEV1 and critical error assessments will be conducted in select patient subgroups only. CAT: COPD assessment test; COPD: chronic obstructive pulmonary disease; FEV1: forced expiratory volume in 1 s; ICS: inhaled corticosteroid; LABA: long-acting β2-agonist; LAMA: long-acting muscarinic antagonist; R: randomisation. #: ICS/LAMA/LABA multiple-inhaler triple therapy, LAMA/LABA combination dual therapy or ICS/LABA combination dual therapy.
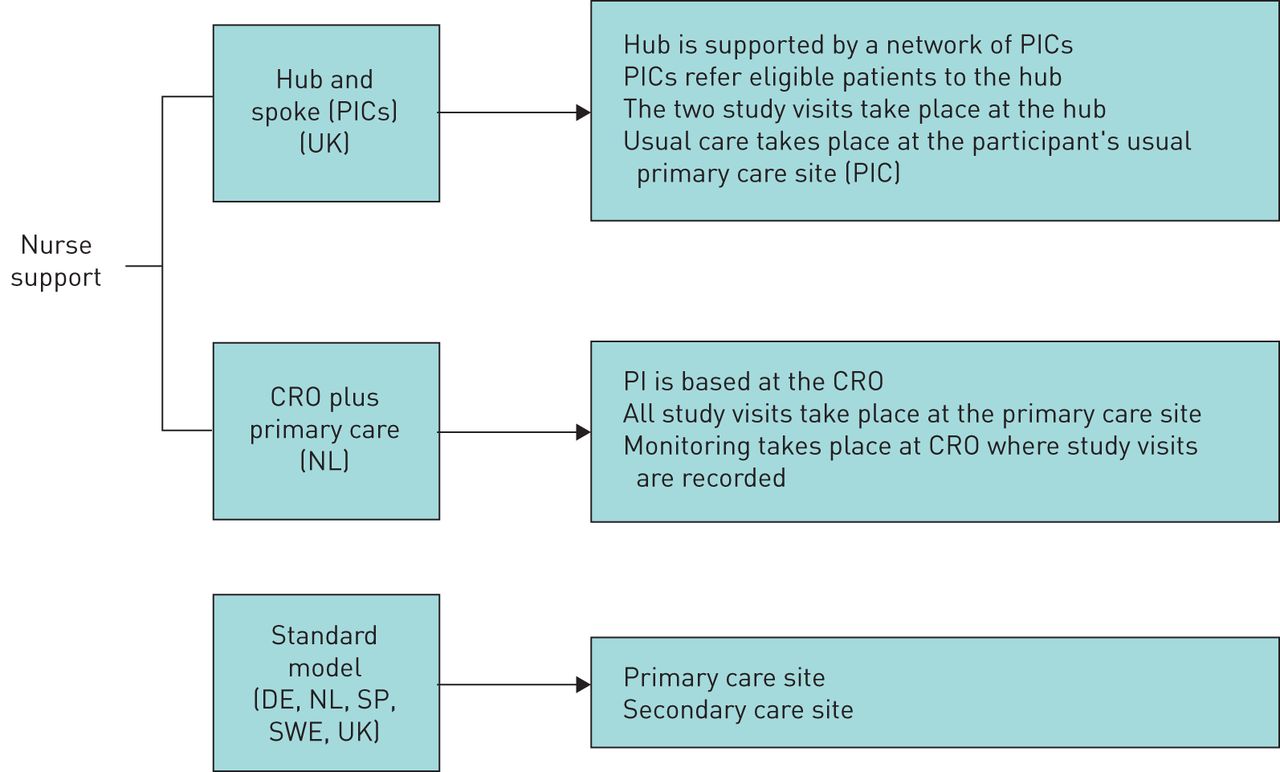
Individual operating models were developed for each country to support reliable data collection while maintaining usual care. To overcome the resource constraints of primary healthcare providers in the UK, a hub and spoke model has been established (figure 2). Patients are identified at primary healthcare sites and then recruited by a regional hub. The hub site may be a secondary healthcare site, but is more often a large general practitioner (GP) surgery. Visits will be conducted by the investigator at selected hub sites and usual care will be provided by the patient's GP between visits.
{kind=link}
{kind=link}
Recruitment models employed across countries. CRO: contract research organisation; DE: Germany; NL: the Netherlands; PI: principal investigator; PIC: patient identification centre; SP: Spain; SWE: Sweden; UK: United Kingdom.
A similar model was established in the Netherlands, with an academic research centre providing the hub services. In Germany and Spain, usual care for the study population is provided by a pulmonologist. For this reason, the patients will see the investigator at both study visits, with no protocol-defined visits in between. When country regulators permit investigational product to be delivered (as in Germany, the Netherlands, Sweden and the UK), participants are given a prescription for their study treatment that is later reimbursed. Participants may then collect their study treatment from any pharmacy, reinforcing usual behaviour. Features of the INTREPID study design allowing the assessment of everyday effectiveness are summarised in box 1.
Patients may augment study treatment with COPD maintenance therapy (excluding FF/UMEC/VI for non-ELLIPTA MITT) and remain in the study. Treatment changes are initiated at the discretion of the investigator or treating physician. For patients randomised to FF/UMEC/VI, the preferred switch will be to any MITT or non-ELLIPTA SITT. For those randomised to non-ELLIPTA MITT, the preferred switch will be to a non-ELLIPTA COPD maintenance therapy, excluding non-ELLIPTA SITT.
This study will be conducted in accordance with applicable local regulations, and the principles stated in the Declaration of Helsinki and the International Conference on Harmonisation Tripartite Guideline for Good Clinical Practice. All study documents will be reviewed and approved by Institutional Review Boards and/or Independent Ethics Committee(s) at all investigational sites. All patients will be asked to provide written, informed consent.
Patient selection
Patients aged ≥40 years, with a physician diagnosis of COPD and a COPD assessment test (CAT) score of ≥10 at screening, who have been receiving a non-ELLIPTA maintenance therapy (ICS/LAMA/LABA MITT, LAMA/LABA combination dual therapy or ICS/LABA combination dual therapy) for ≥16 weeks prior to randomisation and who have a history of ≥1 COPD exacerbation requiring treatment with systemic or oral corticosteroids, antibiotics, and/or hospitalisation in the 3 years prior to randomisation will be eligible for enrolment. Patients enrolled will already be taking triple therapy for COPD or, in the opinion of their physician, will have a clinical need to be stepped up from dual to triple therapy. Key inclusion and exclusion criteria are shown in table 2.
End-points and assessments
The primary end-point will be the proportion of responders based on change from baseline in CAT score at week 24; the CAT is a validated, eight-item, self-completed questionnaire scored on a 40-point scale (patients rate their experience from 0 to 5 for each question), with higher scores indicating greater disease impact [35]. A CAT score reduction of ≥2 units from baseline will be defined as a clinically meaningful response [36]. Change in CAT score was chosen as this outcome measure is utilised in usual practice, is meaningful to patients, informs management of patients by healthcare professionals (HCPs), and is of significant interest to health technology agencies and payers.
Secondary end-points include assessment of lung function (forced expiratory volume in 1 s (FEV1)) and device errors at week 24, both assessed in a subset of patients. Lung function will be assessed by spirometry with centralised quality review prior to study treatment and, if the patient is able to do so, after withholding short-acting β-agonists or short-acting anticholinergics for ≥4 h. Lung function data will be included for patients regardless of their choice to withhold. Standardised spirometry equipment meeting the minimal performance recommendations of Miller [37] will be provided to all sites and used per vendor guidelines. A minimum of three spirometry measures will be obtained and the largest acceptable efforts will be recorded.
There is no central oversight of inhaler training either for HCPs or patients. Patients will be trained on the correct use of their device(s) according to usual clinical practice. For patients in the spirometry subgroup, critical errors, defined as device errors that lead to no or significantly reduced medication being inhaled, and overall errors, defined as any other error in device use [38], will be assessed at the final study visit (week 24). Error checklists will be provided to investigators for scoring correct use; checklists were produced based on a review of the patient information leaflet for each inhaler and the steps defined for correct use, the available literature and a review of these errors with a group of external inhaler experts [39]. Patients will be asked to demonstrate correct inhaler technique when taking their regular medication dose in front of an investigator. Any errors made and instances of correct use will be recorded. For MITT, patients will be tested on each inhaler consecutively. This end-point was included due to the importance of inhaler technique in maximising treatment efficacy [28].
Other clinical end-points will include: a descriptive analysis of the annualised rate of moderate or severe COPD exacerbations (defined as any worsening of symptoms requiring systemic corticosteroids and/or antibiotics or hospitalisation), time to first moderate or severe COPD exacerbation, and clinically important deterioration (defined as 100 mL reduction from baseline in FEV1 at 24 weeks, an exacerbation requiring treatment with antibiotics and/or systemic steroids or hospitalisation, or a ≥2-unit increase from baseline in CAT score).
Safety data will be recorded in the eCRF by the investigator at scheduled and unscheduled visits for COPD-related care on all adverse events (AEs), serious AEs (SAEs) and treatment-related AEs leading to withdrawal from study treatment. AEs not related to study treatment or withdrawal from study treatment will not be recorded unless classified as SAEs; however, all cardiovascular and pneumonia events will be recorded.
In addition to the CAT, participants will complete a short, generic health-related quality of life (HRQoL) questionnaire at baseline, and both a treatment and study satisfaction questionnaire and a HRQoL questionnaire at week 24. The HRQoL questionnaire is a modified version of that used by the Centres for Disease Control and Prevention (online supplement). HRU data will be collected in the eCRF through patient recall and discharge notes. In the UK, the Netherlands and Sweden, COPD HRU data will also be collected directly from national electronic healthcare records.
Planned statistical analyses
For sample size calculations in INTREPID, a conservative odds ratio of 1.3 was assumed for the proportion of CAT responders for the comparison of FF/UMEC/VI versus non-ELLIPTA MITT based on the outcomes of two recent studies: SLS COPD [18], which contained a cohort of patients receiving ICS/LAMA/LABA at baseline who subsequently received FF/VI plus LAMA or usual care, and a further RCT of ELLIPTA SITT versus non-ELLIPTA dual therapy [25].
The null hypothesis for INTREPID is that there will be no difference in the proportion of CAT responders between FF/UMEC/VI and non-ELLIPTA MITT at week 24. Assuming the percentage of CAT responders in the non-ELLIPTA MITT arm is 35% and the true odds ratio between MITT and FF/UMEC/VI is 1.3, 1297 patients per arm would provide 90% power to reject the null hypothesis at the two-sided 5% significance level. Considering the low dropout rate observed in both SLS COPD [18] and the RCT, the dropout rate in INTREPID is assumed to be approximately 13.5%. Thus, a total of 3000 patients will be required for randomisation. All randomised participants will be analysed in their randomised group (intent-to-treat (ITT) population), irrespective of compliance with the trial protocol. The ITT population will be used for the analysis of primary and secondary end-points, which we believe will provide a robust approach to the analysis. Further information will be included in the INTREPID study results manuscript.
For the primary end-point of proportion of CAT responders at week 24, a logistic regression model is planned, with treatment as an explanatory variable and including covariates of baseline CAT score, number of exacerbations in the prior year, prior medication use strata and country. The final statistical analyses will be defined in the reporting and analysis plan. Additional analyses (such as exploratory or regional subgroup analyses) will be defined in the supplementary analysis plan. These will be finalised prior to database lock, and the study protocol and participant-level dataset will be made publicly available following study completion.
For the primary outcome of proportion of CAT responders at week 24, a primary estimand is defined that includes a strategy to handle intercurrent events and missing data. If a participant modifies the treatment they were assigned at randomisation, they will be considered a nonresponder regardless of whether CAT data are available at visit 2. Otherwise, if a participant has missing data at visit 2, multiple imputation methods for values based on the randomised treatment arm characteristics and assuming missing at random will be applied. More details around this approach will be included in the INTREPID study results manuscript.
INTREPID: the benefits and challenges
Randomised effectiveness studies such as INTREPID offer the opportunity to better understand the true benefits of a medicine versus existing standard of care in usual care settings. However, while conceptually simple, the design and implementation of effectiveness studies is complex [40]. INTREPID was designed to provide results with a relatively high degree of both internal and external validity. Internal validity is assured through patient randomisation to parallel study arms, training of investigators and rigorous processes for data management, data analysis and safety follow-up. External validity (generalisability) of the results is then maintained by the alignment of patient care with usual practice in each country; patient randomisation represents the most notable departure from usual care.
INTREPID's setting across multiple geographies and healthcare settings is a particular strength of the study, which represents the first head-to-head comparison of MITT with a single-inhaler approach. It is, nevertheless, challenging to design a study that remains aligned to routine care in all countries of interest.
Minimally interventional end-points were selected based on the balancing of which clinical end-points are collected in usual practice, their interest to physicians, patients and health technology agencies, and their operational feasibility. For example, while exacerbation frequency is an important measure of disease management, the number of patients that would provide adequate statistical power to demonstrate a significant difference in the ability of FF/UMEC/VI and MITT to reduce exacerbation frequency was prohibitively high. Conversely, the collection of HRQoL data is entirely feasible; HRQoL data are key to physicians, patients and health technology agencies alike.
Complexities, such as those associated with the open-label nature of the study, aligning with usual care across geographies and switching between usual care options and FF/UMEC/VI were carefully considered for their potential impact on the sample size needed, and also to ensure that these design elements could be adequately addressed in data analysis and interpretation.
Conclusion
The efficacy–effectiveness gap is driven by medicine characteristics impacted by factors relevant to usual practice that are not present or assessed in conventional RCTs. The INTREPID study will evaluate for the first time whether FF/UMEC/VI is more effective than non-ELLIPTA MITT in usual practice. In the case of FF/UMEC/VI, any differences in efficacy and effectiveness are likely to be driven by differences in the patient populations included in RCTs compared with the patients prescribed FF/UMEC/VI in usual practice, and also by the potential increase in adherence offered by introducing simple, single inhalers compared with the multiple-inhaler regimens currently recommended in usual care.
Box 1 Features of the INTREPID study design that allow the assessment of everyday effectiveness
Broad inclusion criteria reflecting the indication of interest.
Minimal exclusion criteria.
Open-label design.
Commercial medicine supply provided through usual prescription and pharmacy channels where possible.
Country operating models aligned with usual practice.
Patients treated by their usual practice physician between study-specific visits.
Inclusion of only two study visits, allowing minimal impact on patient behaviour.
Protocol allows clinicians to optimise therapy with nonstudy medications.
Primary end-point is a patient-reported outcome that is easy for patients to complete and is used in clinical practice; this end-point is relevant to patients, clinicians and payers.
Acknowledgements
Trademarks are the property of their respective owners. The authors would like to thank both Dimitra Brintziki (GlaxoSmithKline plc., Stockley Park, UK) and Melanie Schroeder (GlaxoSmithKline plc., Brentford, UK) for their contributions to development of the INTREPID study protocol. Editorial support (in the form of writing assistance, collating author comments, assembling tables/figures, grammatical editing, fact checking and referencing) was provided by Matthew Hallam of Gardiner-Caldwell Communications (Macclesfield, UK) and was funded by GlaxoSmithKline plc.
Footnotes
This article has supplementary material available from openres.ersjournals.com
This study is registered at www.clinicaltrials.gov with identifier number NCT03467425. Study documents can be requested for further research from www.clinicalstudydatarequest.com.
Author contributions: A.S. Ismaila performed the literature search. D. Midwinter, M. Tabberer, A.S. Ismaila, L. Sansbury, D.M.G. Halpin, D. Leather, E. Irving, S. Worsley and N. Snowise designed the study. All authors interpreted data, wrote and reviewed the manuscript, and gave final approval of the manuscript.
Support statement: This study was funded by GlaxoSmithKline plc. (GlaxoSmithKline plc. study no. 206854; ClinicalTrials.gov identifier: NCT03467425).
Conflict of interest: S. Worsley is an employee of, and holds stock in, GlaxoSmithKline plc.
Conflict of interest: N. Snowise is an employee of, and holds stock in, GlaxoSmithKline plc., holds shares in Vectura, and is a Visiting Senior Lecturer at the Faculty of Life Sciences and Medicine, King's College, London.
Conflict of interest: D.M.G. Halpin reports personal fees from AstraZeneca, Chiesi, GlaxoSmithKline plc. and Pfizer and personal fees and nonfinancial support from Boehringer Ingelheim and Novartis, outside the submitted work.
Conflict of interest: D. Midwinter is an employee of, and holds stock in, GlaxoSmithKline plc.
Conflict of interest: A.S. Ismaila is a full-time employee of GlaxoSmithKline plc. and an unpaid, part-time professor at McMaster University.
Conflict of interest: E. Irving holds stocks and shares in GlaxoSmithKline plc. R&D Ltd.
Conflict of interest: L. Sansbury is an employee of, and holds stock in, GlaxoSmithKline plc.
Conflict of interest: M. Tabberer is an employee of, and holds stock in, GlaxoSmithKline plc.
Conflict of interest: D. Leather is an employee of, and holds stock in, GlaxoSmithKline plc.
Conflict of interest: C. Compton is an employee of, and holds stock in, GlaxoSmithKline plc.
- Received March 4, 2019.
- Accepted September 2, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.