



Matrix metalloproteinase-7 is increased in lung bases but not apices in idiopathic pulmonary fibrosis
- Jade Jaffar1,2⇑,
- Mae Wong3,
- Gregory A. Fishbein4,
- Monther Alhamdoosh3,
- Laura McMillan3,
- Cristina Gamell-Fulla3,
- Milica Ng3,
- Nick Wilson3,
- Karen Symons2,
- Ian Glaspole1,2 and
- Glen Westall1,2
- 1Department of Immunology and Pathology, Monash University, Melbourne, VIC, Australia
- 2Department of Respiratory Medicine, The Alfred Hospital, Melbourne, VIC, Australia
- 3CSL Limited, Parkville, VIC, Australia
- 4David Geffen School of Medicine at UCLA, Los Angeles, CA, USA
- Corresponding author: Jade Jaffar (j.jaffar{at}alfred.org.au)
Abstract
Introduction Idiopathic pulmonary fibrosis (IPF) is a progressively fibrotic lung condition with poor prognosis. Matrix metalloproteinase-7 (MMP7) is a protein secreted by epithelial cells in IPF lungs. It is not known if MMP7 expression correlates with fibrotic changes in lung tissue.
Methods Tissue samples from lung apices and bases were obtained from 20 IPF patients and 14 non-diseased control (NDC) donors. In formalin-fixed paraffin-embedded sections, histological assessment of fibrosis was performed; overall MMP7 positivity was assessed by immunohistochemistry and MMP7+ cells were quantified using multiplex immunohistochemistry. Protein expression of MMP7 in whole lung lysates was quantified by Western blotting. Bulk tissue transcriptomic profiles of 101 samples were analysed using RNA sequencing technologies.
Results Lung tissue from IPF bases was more fibrotic than in apices. MMP7 protein is elevated in IPF lung base tissue. In IPF whole lung lysates, MMP7 protein levels are increased compared to NDC donors and was increased in IPF lung bases compared to apices. MMP7 protein levels correlated with MMP7 gene expression levels in lung tissue. MMP7 transcript levels were increased in IPF base compared to NDC base lung tissue and increased in IPF base tissue compared to IPF apex tissue.
Conclusions Our cross-sectional study suggests that lung epithelial MMP7 expression increases as the tissue becomes more fibrotic and identifies a potentially nonepithelial or immune-cell source. Mechanisms of disease progression in IPF are still unclear, and our study suggests aberrant MMP7 production may be a histological starting point of lung tissue fibrosis.
Abstract
This study suggests that lung epithelial MMP7 expression increases as the tissue becomes more fibrotic and identifies a potentially nonepithelial or immune-cell source https://bit.ly/3QsXYFP
Introduction
Idiopathic pulmonary fibrosis (IPF) remains a chronic interstitial lung disease (ILD) with no known cure despite the availability of two anti-fibrotic compounds shown to slow disease progression [1]. With an unknown aetiology [2], development of effective drug treatments for IPF is challenging [1], and additionally, clinically relevant tissue markers of active fibrosis are lacking [3]. Matrix metalloproteinase-7 (MMP7) is a member of a family of 28 calcium-dependent endopeptidases that are crucial in tissue homeostasis and wound healing [4]. MMP7 is a well-known, extensively studied serum biomarker of disease progression in IPF [5–9]. MMP7 knockout mice are protected from bleomycin-induced pulmonary fibrosis [10], highlighting the role MMP7 plays in IPF disease progression. However, it is not known if MMP7 expression changes as tissue becomes more fibrotic and could also be a histological marker of fibrotic disease activity in IPF.
In IPF, fibrotic disease develops in a characteristic geographical pattern in the lung, with the fibrosis typically starting at the bases and progressing to the apices as the disease worsens. The importance of this geospatial polarity was emphasised by McDonough et al. [11] who showed transcriptomic differences between IPF lung tissue which had a more normal histologic structure (pre-terminal stage) compared to more fibrotic IPF lung tissue that was at later stages of disease progression.
We reasoned that due to the basal predominance of disease progression, apical lung tissue sampled at the time of lung transplantation would be significantly less fibrotic than tissue derived from the bases in patients with IPF. Lung apex-derived tissue could therefore represent a pre-terminal stage, whereas base-derived tissue would be more characteristic of “end-stage” fibrosis. Since elevated serum MMP7 levels are predictive of poor prognosis in IPF [5–8], we hypothesised that tissue MMP7 expression would also increase from histologically normal regions to areas of pre-terminal fibrosis, and then to end-stage fibrosis. Therefore, the aim of this study was to investigate the expression of MMP7 in tissue sampled from lung apices and bases in patients with IPF and non-diseased control (NDC) donors and then correlate the levels with tissue representative of pre-terminal and end-stages of fibrosis.
Methods
For detailed methods refer to the supplementary material.
Study subjects
Ethical approval for this study was granted by The Alfred Human Ethics Committee (#468/14) and the Australian Red Cross Blood Service Ethics Committee and met requirements of the Australian National Statement on Ethical Conduct in Human Research (2007). Written, informed consent was obtained from all study participants or from their families.
Explant lung tissue and whole lung lysates were obtained from patients with IPF (n=20). Two patients with IPF were on anti-fibrotic medication prior to lung transplantation. Tissue from NDCs (n=14) were obtained from deceased organ donors whose lungs had been declined for transplantation. Patient demographic information can be found in table 1.
Patient demographics
Study design
Targeted samples were taken from the lung apex and base from each subject and analysed in this study (supplementary figure S1). Details regarding the samples used in each of the experimental datasets can be found in supplementary table S1. Lung tissue from the right and left upper lobes (RUL, LUL) was compared to tissue from the right and left lower lobes (RLL, LLL) from all subjects.
Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) sections were immunohistochemically stained for MMP7 and E-cadherin (epithelial marker), CD31 (endothelial marker), CD45 (myeloid marker) and α-smooth muscle actin (αSMA, mesenchymal marker).
Pathologist brightfield immunohistochemistry assessment of MMP7
MMP7 protein was visualised for brightfield in apex and base matched tissue from 12 patients with IPF and 10 NDC donors. Sequential tissue sections were used for the pathologist's (G.A. Fishbein) assessment of fibrotic stage. Histological extent of fibrosis (Fibrosis Score) in tissue from IPF patients was graded on a scale of 1–4. Additional details regarding staining specificity can be found in the supplementary material.
Multiplex staining
FFPE tissue sections were first stained with Masson's trichrome to assess extent of fibrosis (supplementary figure S2A). Tissue sections from three histologically normal NDC donors and three patients with IPF whose tissue showed distinct differences between the extent of fibrosis in the lung apex compared to the lung base were used for multiplex staining. A total of nine samples were used in the analysis.
For multiplex staining, tyramide signal amplification was performed. To assess potential crossover staining, tissue was stained with individual markers during optimisation of the panel (supplementary figure S2B).
Sections were scanned using the Vectra 3 Quantitative Pathology Imaging System (Perkin Elmer, Hopkinton, MA, USA) and regions of interest were identified for subsequent analysis at 20× magnification (supplementary figure S2C). Quantification of marker positive cells was performed using HALO imaging processing software on a total of 1101 images. Data are presented as percentage of positive cells per total number of cells counted.
Immunoblotting
MMP7 and GAPDH (Abcam, Cambridge, UK) were immuno-detected in lysates of lung tissue. To validate MMP7 antibody specificity and act as positive controls, epithelial A549 cell culture supernatant (ATCC, Manassas, VA, USA) and recombinant MMP7 protein (R&D Systems, Minneapolis, MN, USA) were used. Data are presented as the log of the ratio of zymogen MMP7 to GAPDH bands.
Transcriptome analysis
A total of 105 apex and base samples, from 20 IPF patients and 14 NDC donors, were collected for RNA sequencing (RNA seq) and 101 were used in the analysis (GEO database number GSE213001).
Bioinformatics analysis
A gene was considered differentially expressed if it was statistically significant at false discovery rate (FDR, Type I error) cut-off of 0.05 and fold change of 2.
Statistics
Histological analysis used the Mann–Whitney t-test to compare NDC and IPF samples, while the Wilcoxon paired t-test was used to compare apex and base samples. For Western blots, differences between groups were assessed by one-way ANOVA with Holm–Sidak's test for multiple comparisons. Within group comparisons were performed with paired t-test. Pearson correlation coefficient was calculated between gene and protein MMP7 levels in the bulk tissue experiments, whereas Spearman's rank was calculated for cell counting experiments.
Results
MMP7 expression is increased in end-stage IPF compared to control tissue
The extent of fibrosis in the lung tissue from patients with IPF was greater in samples derived from lung bases (end-stage) compared to matched lung apices (pre-terminal disease) (figure 1b). In tissue from IPF patients, only one of 24 samples had no observable MMP7 expression and were apex-derived. In three other apex-derived IPF lung tissue samples, MMP7 expression was limited to the rare respiratory epithelial cell. Strong MMP7 expression could be seen in elastin fibres of six lung base-derived IPF lung tissue and the other six lung base-derived samples had apical and cytoplasmic MMP7 positivity in respiratory epithelial cells. MMP7 expression was observed in areas of fibrosis in some macrophages and with nonspecific staining of elastin fibres. Figure 1a shows representative MMP7 staining of lung tissue from apex and base of 12 IPF and 10 NDC lungs. MMP7 expression in NDC lung tissue was minimal with four out of 20 samples having rare epithelial cell staining and one sample with nonspecific mucus staining (supplementary table S2).

Matrix metalloproteinase-7 (MMP7) expression is increased in IPF compared to control tissue and greatest in areas of end-stage disease. a) Representative brightfield staining of MMP7 in formalin-fixed paraffin-embedded tissue from the apex and base of 12 patients with idiopathic pulmonary fibrosis (IPF) and 10 non-diseased control (NDC) donors whose lungs were unsuitable for transplantation. A total of 44 sections were analysed (supplementary table S1). Staining specificity was assessed for every sample by omitting the MMP7 antibody on sequential sections. Inserts show regions in IPF tissue at higher resolution. Open arrows show MMP7 positivity in elastin fibres. Filled arrows indicate cytoplasmic and apical staining on respiratory epithelial cells. Scale bars: solid line 500 μm; double line 100 μm. b) From the IPF patients, sequential tissue sections were stained with haemotoxylin and eosin and assessed for extent of fibrosis by a qualified pathologist (G.A. Fishbein) and a Fibrosis Score (1=mild fibrosis, 2=moderate fibrosis/possible usual interstitial pneumonia (UIP), 3=favour UIP or 4=end-stage fibrosis) was applied. Summary of the analysis is found in supplementary table S2. *p=0.0234, Wilcoxon paired t-test. c) Overall MMP7 positivity in each tissue section was graded on the following scale: 1=No or rare staining; 2=subset of positive cells observed/moderate staining; or 3=Yes staining. *p=0.0115 and ****p<0.0001, Mann–Whitney t-test, **p=0.0078 Wilcoxon paired t-test.
MMP7 positivity was also higher in IPF tissue from both apex and base compared to NDC tissue. MMP7 positivity was increased in IPF lung bases compared to matched apices (figure 1c). MMP7 positivity correlated with Fibrosis Score in IPF lung tissue (Pearson R=0.54, p=0.0067, n=24).
MMP7 protein levels are increased in IPF whole lung lysates from bases but not apices compared to control tissue
Figure 2a shows an immunoblot of matched apex- and base-derived whole lung lysates from three IPF patients and three NDC donors probed for MMP7 and the loading control GAPDH. MMP7 was measured in apex and base lung tissue from 12 IPF (11 paired samples) and 11 NDC (11 paired samples) donors. Assay specificity was verified using epithelial A549 cell culture supernatant and recombinant MMP7 protein. While MMP7 levels in whole lung lysates from IPF apices were not different to those from NDC apices, MMP7 levels in IPF bases was higher compared to NDC bases (figure 2b). There was no difference in MMP7 level between NDC apices and bases, but MMP7 was increased in IPF bases compared to apices (figure 2b). The full set of immunoblots with lung locations can be found in supplementary figure S2.

Matrix metalloproteinase-7 (MMP7) protein levels are increased in IPF whole lung lysates from bases but not apices compared to control tissue. MMP7 levels in whole lung lysates of 12 idiopathic pulmonary fibrosis (IPF) patients and 11 non-diseased control (NDC) donors were quantified by immunoblot. a) A representative immunoblot showing matched (apex and base) samples from N=3 IPF and N=3 NDC donors is shown. MMP7 zymogen (inactive) is detected at 28 kDa (green bands) and GAPDH (red bands, 36 kDa). Culture supernatant from A549 epithelial cells and activated recombinant MMP7 (18 kDa) are shown as positive controls. b) The level of MMP7 zymogen relative to GAPDH was significantly higher in IPF apex tissue compared to IPF base tissue (n=11 pairs). The level of MMP7/GAPDH was elevated in IPF base (n=12 samples) compared to NDC base (n=11 samples). Between-group comparisons were performed with one-way ANOVA with Holm–Sidak's test for multiple comparisons. Within group comparison was performed with paired-t-test. One IPF base sample without an apex match is shown as a half-filled in square.
MMP7 gene expression correlated with MMP7 protein levels and was highest in end-stage tissue
Whole transcriptome of bulk apex and base tissue from 20 IPF patients and 14 NDC donors was profiled using RNA seq (supplementary table S1). In 45 samples from 12 IPF and 11 NDC donors matched to the RNA seq data (supplementary table S1), MMP7 gene levels correlated with MMP7 protein levels in whole lung lysates (figure 3a). In 44 samples from 12 IPF and 10 NDC donors matched to the RNA seq data (supplementary table S1), MMP7 gene levels in histologically end-stage tissue were higher than in tissue with histologically mild fibrosis (figure 3b). There was no difference in MMP7 gene expression between NDC apex- and base-derived tissue (figure 3c). MMP7 gene expression was 4.92× higher in apex-derived IPF tissue compared to NDC apex-derived tissue, but the difference was not statistically significant (FDR=0.39). MMP7 gene expression was 28× higher in base-derived IPF tissue compared to NDC base-derived tissue (figure 3c).

Matrix metalloproteinase-7 (MMP7) gene expression correlated with MMP7 protein levels and was highest in end-stage tissue. a) MMP7 gene levels and MMP7 protein levels correlated in 45 samples of lung tissue from the lung apex (open symbols) and base (closed symbols) of 12 patients with idiopathic pulmonary fibrosis (IPF, square symbols) and 11 non-diseased control (NDC, circle symbols) donors (Pearson R=0.521, 95% confidence interval 0.27–0.71). b) Matched lung tissue was assessed for both extent of fibrosis via a trained pathologist (G.A. Fishbein) and MMP7 gene levels using RNA seq in 12 IPF patients. Values for extent of fibrosis scoring are found in supplementary table S2. *p=0.272, Kruskal–Wallis one-way ANOVA with Dunn's multiple comparisons test. c) MMP7 gene expression levels in bulk lung tissue from the apex and base of 20 IPF patients and 14 NDC donors was assessed by RNA seq. *: FDR <0.05; ****: FDR ≤0.0001. UIP: interstitial pneumonia; FC: fold change to first group mentioned; FDR: false discovery rate; NS: nonsignificant; RPKM: reads per kilobase million.
MMP7+ epithelial cells are increased and correlated with MMP7 gene expression in IPF lung tissue
Apex-derived lung tissue from three NDC donors and matched apex- and base-derived tissue from three IPF patients whose apex tissue was clearly less fibrotic than their base tissue (supplementary figure 3A) was stained simultaneously for MMP7, E-cadherin, CD31, CD45 and αSMA. Cell nuclei were marked with Hoechst dye. Representative composite images are shown in figure 4. In NDC tissue, MMP7 expression (arrows) was rare and when observed, localised to the alveolar walls. MMP7 expression was occasionally observed in cells that expressed neither E-cadherin nor CD45 (or CD31 or αSMA). In IPF base tissue, MMP7 expression was prominent in the hyperplastic epithelium and in honeycomb cysts.
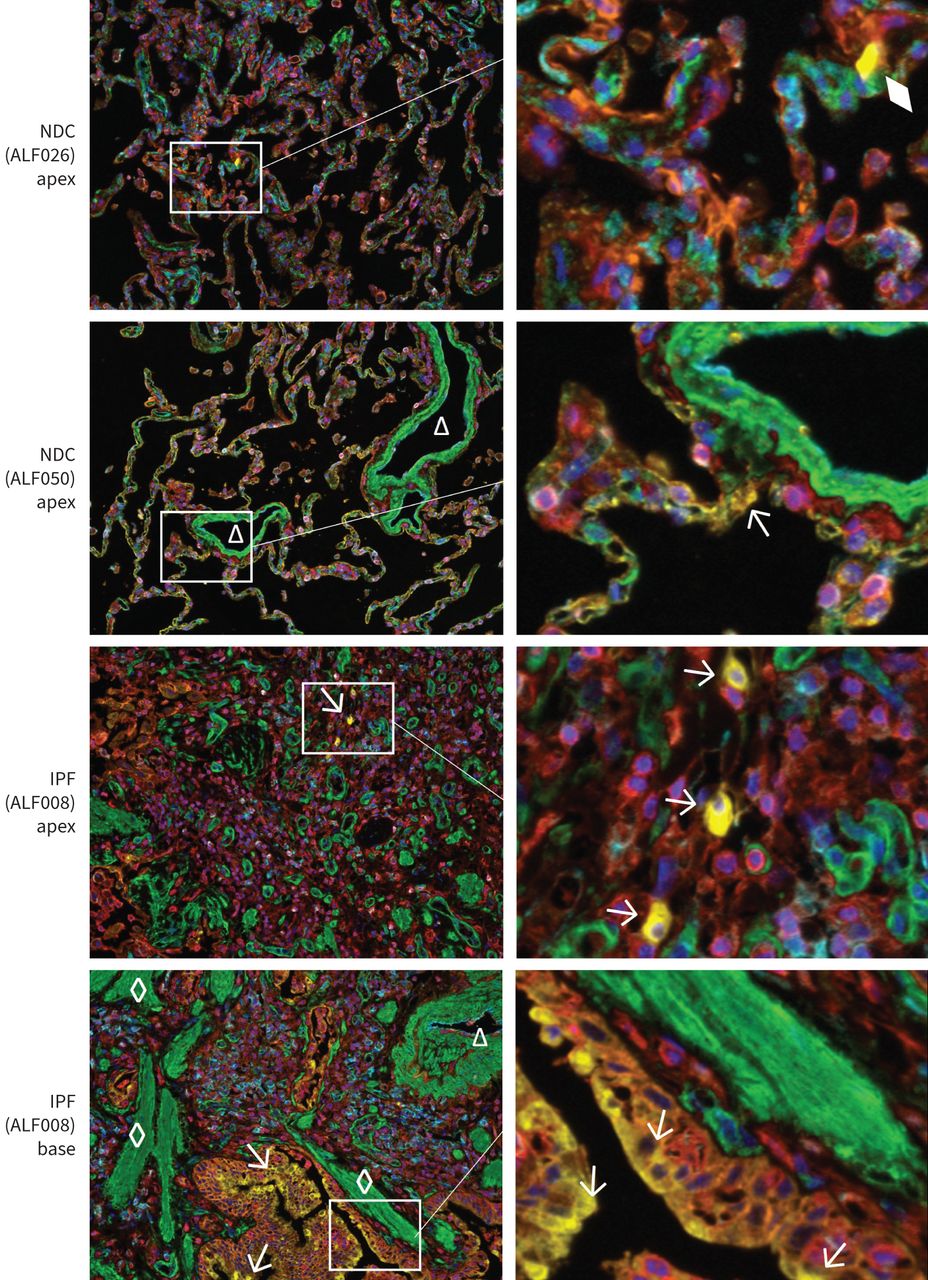
Multiplex immunohistochemistry of matrix metalloproteinase-7 (MMP7). Representative images of idiopathic pulmonary fibrosis (IPF) and non-diseased control (NDC) tissue stained simultaneously for MMP7 (pseudocoloured yellow), E-cadherin (orange), CD45 (red), CD31 (cyan) and α-smooth muscle actin (SMA, green). Cell nuclei are counterstained with Hoechst (blue). Left panels show tissue imaged at 20× magnification captured with the Vectra 3 Quantitative Pathology Imaging System. Triangles indicate blood vessels, diamonds mark fibroblastic foci and arrows point to MMP7+ cells. The diamond shows a non-cellular form (no nucleus) that was not considered an MMP7+ cell.
Overall, 697 457 cells were analysed and 13 933 MMP7+ cells (2.0%) were identified. The numbers of MMP7+ cells in NDC, IPF apex or IPF base tissue were not statistically significant between groups (p>0.05) (figure 5a). The main cells expressing MMP7 protein were E-cadherin positive, averaging 84% of the number of MMP7+ cells counted in NDC tissue, 59% in IPF apex and 81% in IPF base tissue. The number of MMP7+/E-cadherin+ cells counted in NDC lung tissue averaged 41 (0.08% of the total number of cells counted), 638 (0.89%) in IPF apex tissue and 2977 (2.82%) in IPF base tissue (figure 5b). The number of MMP7+/E-cadherin−/CD45−/CD31−/αSMA− cells averaged 0 in NDC tissue, 93 (0.14%) in IPF apex tissue and 407 (0.35%) in IPF base tissue (figure 5c).

Matrix metalloproteinase-7 (MMP7+) epithelial cells are increased and correlated with MMP7 gene expression in IPF lung tissue. Formalin-fixed paraffin-embedded (FFPE) lung tissue from the apex and base of three patients with idiopathic pulmonary fibrosis (IPF) and from the apex of three non-diseased controls were stained sequentially with antibodies specific for MMP7, E-cadherin, CD45, CD31 and α-smooth muscle actin. Nuclei were stained with Hoescht dye. Positive staining was visualised with OPAL dyes (Perkin Elmer) and images were captured at 20× magnification using the Vectra 3 Quantitative Pathology Imaging System. a) The number of MMP7+ cells, b) MMP7+/E-cadherin+ and c) MMP7+/E-cadherin−/CD45−/CD31−/α-smooth muscle actin− (elseNeg) cells are presented as a percentage of the total number of cells analysed. There was no difference between the groups. Absolute numbers of phenotyped cells can be found in supplementary figure S4D. The MMP7 gene expression level in matched samples correlates with d) the percentage of MMP7+ cells (Spearman R=0.74, p=0.0268), e) the percentage of MMP7+/E-cadherin cells+ (Spearman's R=0.73, p=0.0311) and f) percentage of MMP7+/elseNeg cells (Spearman's R=0.83, p=0.0093). There was no difference in g) total number of cells quantified, h) mean area analysed or i) the percentage of E-cadherin+ cells between the groups.
The percentage of MMP7+ and MMP7+/E-cadherin+ cells correlated with MMP7 gene expression levels (figure 5d, e). The percentage of MMP7+/E-cadherin−/CD45−/CD31−/αSMA− cells also correlated with MMP7 gene expression (figure 5f). There was no significance difference in total cells counted (figure 5g), area analysed (figure 5h) or percentage of E-cadherin+ cells (figure 5i) between the groups.
The epithelial transcriptomic profile of IPF apex tissue is significantly different compared to IPF base tissue and can closely resemble NDC tissue
In cluster analyses, there was a clear separation of samples derived from IPF and NDC donors (figure 6a). When comparing samples derived from apex and base, cluster analyses demonstrated that separation between apex and base samples occurred in IPF but not NDC donors (figure 6b). There was no difference between samples derived from left or right lobes in IPF or NDC groups (figure 6c). Figure 6d summarises the 15 065 genes identified by RNA sequencing that we considered differentially expressed (DEGs) between IPF apex, IPF base, NDC apex and NDC base tissue. When comparing IPF apex to IPF base tissue, 920 genes were DEG. Comparing NDC apex with IPF apex tissue resulted in 1490 DEGs, while comparing NDC base with IPF base tissue resulted in 1870 DEGs. There were 248 DEGs between NDC apex and NDC base tissue. There were 2954 unique DEGs when comparing 1) NDC apex versus IPF apex, 2) NDC base versus IPF base and 3) IPF apex versus IPF apex. Of these, only 29 were shared between the three comparisons. 1253 (92%) of the DEGs from 1) NDC apex versus IPF apex and 1285 (80%) of the DEGs from 2) NDC base versus IPF base were shared with other groups compared to only 85 (28%) of DEGs from 3) IPF apex versus IPF base comparisons. 835 (72%) DEGs were unique to the (3) IPF apex versus IPF base comparison. A summary of the detected DEGs at various fold change cut-offs is found in supplementary table S3.

Cluster analysis and common differentially expressed genes of the RNA sequencing dataset. A total of 15 065 genes were identified in 101 lung apex and base tissue samples by RNA sequencing. A gene was considered differentially expressed if it had a false discovery rate (Type I error) of <0.05 and had a fold change (FC) of >log2(1). A summary of the samples and lung locations included in the analysis is found in supplementary table S1. a) Cluster analyses were carried out to compare the overall transcriptomic profile between NDC (blue, n=47 samples) and IPF (red, n=58 samples). There was a clear separation between these two groups. b) When comparing apex and base samples from IPF and NDC donors, cluster analysis showed that there was only a separation between apex and base samples from IPF (n=28 apex, n=29 base) but not NDC (n=23 apex, n=23 base) donors. c) When comparing left and right samples from IPF and NDC donors, cluster analysis demonstrated that there was no separation between samples from the left compared to right lobes in IPF (n=31 left, n=26 right) or NDC (n=22 left, n=22 right) donors. d) The majority (59%) of differentially expressed genes (DEGs) between NDC apex versus IPF apex and NDC base versus IPF base were shared between the two comparisons (1241 shared DEGs compared to 2119 DEGs in total). Conversely, among the 902 DEGs between IPF apex and IPF base, only 85 (28%) were shared with either NDC apex versus IPF apex or NDC base versus IPF base. 29 of 2954 unique DEGs (0.98%) were shared between all three comparisons. IPF: idiopathic pulmonary fibrosis; NDC: non-diseased control.
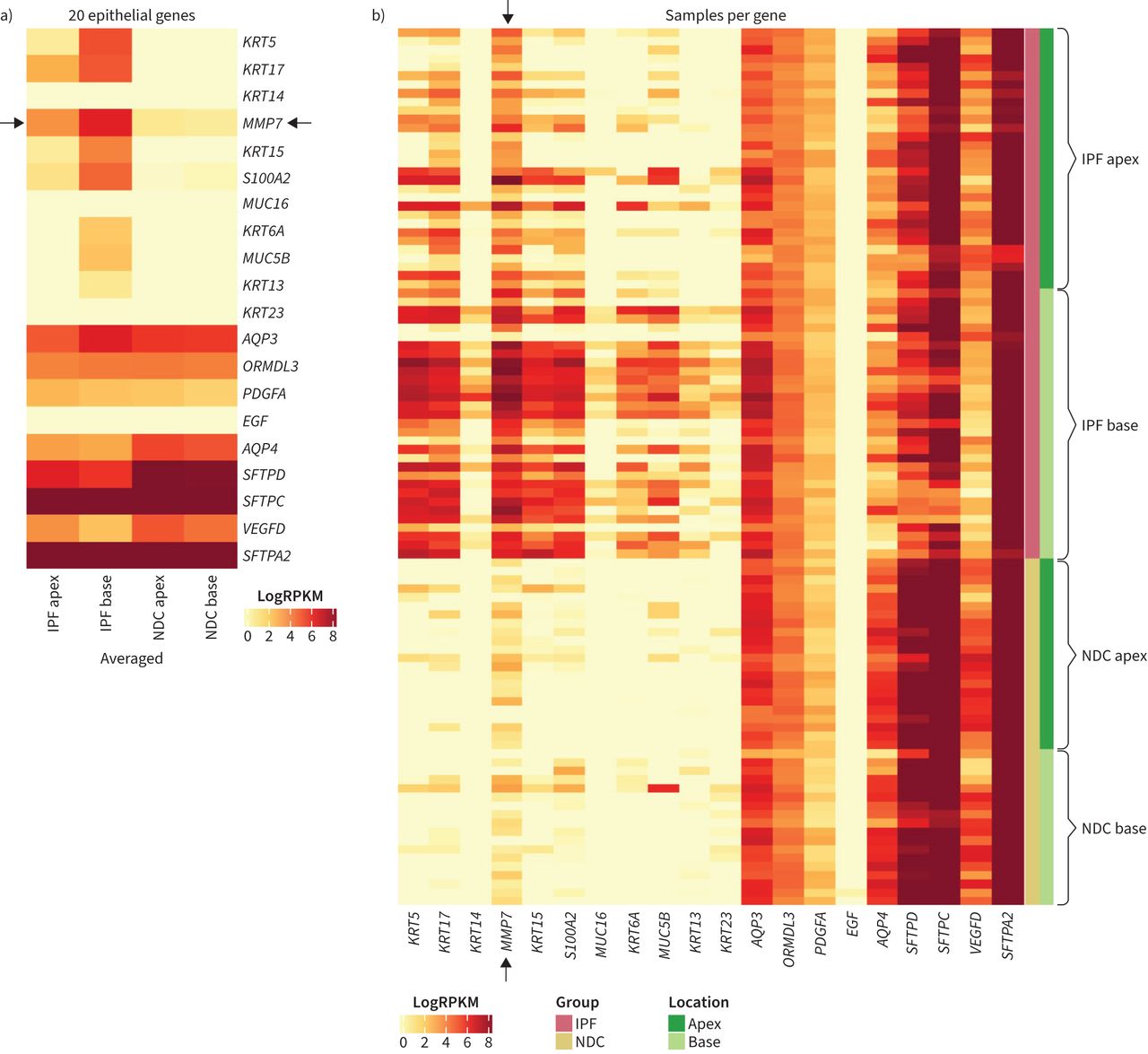
In addition to MMP7, the levels of 19 other epithelial-related genes between were compared in IPF apex, IPF base, NDC apex and NDC base tissue. In the differential gene expression analysis, aquaporins (AQP3 and AQP4), keratins (KRT5, KRT6A, KRT13, KRT14, KRT15, KRT17, KRT23), mucins (MUC16, MUC5B), surfactants (SFTPA2, SFTPC, SFTPD), growth factors (EGF, PDGFA, VEGFD) and S100A2 were differentially expressed in IPF bases compared to IPF apices. The gene ORMDL3 was not differentially expressed between IPF bases compared to IPF apices. Heat maps of the levels of these 20 pre-selected genes in lung tissue are presented in figure 7a and show that the selected gene expression profile is very similar between NDC apex and base tissue, but not between IPF apex and base tissue. Based on their pattern of expression, this set of epithelial genes could be separated into three categories: 1) 11 genes that were increased in IPF base compared to IPF apex and NDC tissue (KRT5, KRT17, KRT14, MMP7, KRT15, S100A2, MUC16, KRT6A, MUC5B, and KRT13); 2) six genes that were decreased in IPF compared to NDC tissue regardless of lung location (EGF, AQP4, SFTPD, SFTPC, VEGFD and SFTPA2); and 3) three genes that were less different between IPF compared to NDC tissue (AQP3 and PDGFA). Compared to NDC apex tissue, 8 out of 20 genes had a log fold change >2 in IPF apex tissue and 14 out of 20 genes in IPF base tissue (figure 7b). The six genes in IPF base but not IPF apex tissue that had a log fold change >2 compared to NDC apex tissue were KRT15, S100A2, MUC16, AQP4, SFTPD and VEGFD. None of the 20 genes had a log fold change >2 in NDC base compared to NDC apex tissue. ORMDL3 was not different between the groups. In differential gene expression analysis, when all IPF samples were compared to all NDC samples, none of the 20 genes were DEG. The mean log fold change in gene expression of the 20 epithelial genes can be found in supplementary table S4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The epithelial transcriptomic profile of IPF lung bases is significantly different to apices. Matrix metalloproteinase-7 (MMP7) and 19 other epithelial genes were pre-selected for comparison. Heatmaps showing the a) averaged and b) individual expression levels of the 20 epithelial-related genes. Black arrows denote MMP7 bands. The log fold changes of the 20 genes in NDC base, IPF apex and IPF base tissue compared to NDC apex can be found in supplementary table S3. IPF: idiopathic pulmonary fibrosis; NDC: non-diseased control; RPKM: reads per kilobase million.
Discussion
Studies investigating the spatial heterogeneity of fibrosis in IPF are lacking. Our study uses a selection of approaches to probe the spatial expression of MMP7 between the upper and lower lobes in patients with IPF. This study is also the first to observe a population of strongly MMP7-expressing cells in IPF tissue that did not express E-cadherin, αSMA, CD31 or CD45, markers typical of epithelial, mesenchymal, endothelial or immune cells respectively. These MMP7+ cells were present in apex-derived IPF tissue (e.g. pre-terminal stage) which was more histologically normal and had an epithelial transcriptomic profile more like normal tissue than matched tissue at the end-stages of fibrosis. MMP7 gene expression in IPF apex tissue was not elevated compared to NDC tissue and was decreased compared to matched IPF base tissue (e.g. end-stage), implying that MMP7-expressing cells may be important during the later stages of fibrosis. This study also demonstrated that protein levels of MMP7 correlated with gene levels in matched bulk tissue analyses, suggesting that significantly elevated gene expression of MMP7 may be indicative of concurrent pathological enzymatic processes occurring in lung tissue.
Data from the IPF Cell Atlas (https://doi.org/10.1152/ajplung.00451.2020) shows that the highest MMP7-expressing cells in IPF are aberrant basaloid, club and goblet cells which all also express E-cadherin. This leads us to speculate that one mechanism of disease progression in IPF involves MMP7 secretion into the interstitial space by damaged epithelial cells. Single-cell studies have also described an “aberrant basaloid” cell in IPF lung tissue that expressed markers of airway basal cells as well as mesenchymal markers (COL1A1) and MMP7 [12].
Honeycomb formation and fibroblastic foci at the leading edge of fibrotic tissue destruction make up the distinct histological pattern of IPF called usual interstitial pneumonia (UIP) [13]. In an attempt to explain the pathophysiology of IPF, Snijder et al. [14] conclude that honeycombing represents a progressive enlargement of the distal airways and involves epithelial cell dysfunction. In our study, MMP7 gene expression was elevated in IPF base versus NDC base tissue but was not elevated in IPF apex compared to NDC apex tissue. MMP7 protein levels were similarly increased in IPF bases but not apices. Therefore, we postulate that excessive MMP7 production occurs only after pathogenic remodelling of IPF lung tissue has already started.
The expression of MMP7 is regulated by the Wnt/β-catenin pathway, which is known to play a role in IPF disease pathogenesis, particularly within the respiratory epithelium [15]. In lung epithelial cells, MMP7 regulates β-catenin to promote wound healing and tissue remodelling [16] and is therefore necessary for homeostatic maintenance of tissue [17]. However, pathological epithelial cell dysfunction may occur later in the fibrogenic process, perhaps because of a dysregulated and fibrotic extracellular matrix (ECM), which is known to drive IPF pathology [18–21]. It is also possible that there are fibroblast-like cells which express MMP7 that drives honeycomb cyst formation.
MMP7 is secreted as an inactive enzyme containing two conserved motifs, the pro-domain and the catalytic domain [22]. Upon cleavage of the pro-domain, the catalytic domain can then bind specific substrate molecules, which in the case of MMP7 include E-cadherin and Fas ligand [16]. MMP7 can also bind ECM proteins such as fibronectin, elastin, type IV collagen and proteoglycans as well as other MMPs such as MMP2 and MMP9 [23]. MMP7 activation is controlled through substrates that contain highly sulfated glycosaminoglycans (GAGs) such as heparin [24]. The observation in our study that activated MMP7 was only detected in whole lung lysates from IPF lung bases suggests that aberrant activation of MMP7 may be involved in disease progression during end-stage fibrosis. In the context of pancreatic cancer cells, MMP7 can be activated by plasmin [25], which we have previously shown to play a role in the pro-fibrotic activation of lung fibroblasts [26]. In that study, lung fibroblasts from patients with IPF produced more plasmin than those from control donors, so activation of MMP7 by plasmin may be another mechanism driving dysregulated tissue remodelling in IPF. Further investigation into the cross-talk between MMP7-producing activated fibroblasts and epithelial cells is warranted.
Although the precise mechanism driving IPF remains undefined, damage to the epithelium is known to play an important role [27]. A 2016 study using single-cell RNA sequencing identified distinct subsets of epithelial cell types that “shared characteristics of conducting airway basal and goblet cells and an additional transitional cell that contributes to pathological processes in IPF” [28]. Since the greatest level of transcriptomic activity occurred in IPF bases where end-stage honeycomb cyst formation and fibroblastic foci activity are greatest, perhaps the permanently altered tissue architecture may not be totally static and/or something about the end-stage fibrotic micro-environment drives heightened cellular activity. Importantly, none of the 20 pre-selected epithelial-related genes were differentially expressed when IPF and NDC samples were compared regardless of lung location, and only SFTPA2 was differentially expressed between IPF apices and NDC apices.
Keratins are a large family of proteins involved in epithelial cell homeostasis [29]. Mucins are epithelial-produced key effectors of cell growth and tissue remodelling that are known to play a role in pulmonary fibrosis [30]. Alveolar surfactants are epithelial Type II cell-produced lipids required for normal respiration [31]. Mutations in surfactant genes have been identified in IPF [32]. Aquaporins are channel proteins in the membranes of cells tasked with regulating water transport between them [33]. Elevated expression of aquaporin gene AQP3 has been reported in IPF [28]. Epidermal growth factor (EGF) levels are increased in patients with pulmonary hypertension [34], and increased EGF receptor is associated with IPF [35]. Signalling through VEGF-D (and VEGF-C) is a central mechanism of fibrosis-related lymphangiogenesis [36]. These epithelial genes may reflect the specific micro-environment of end-stage fibrosis. Taken together, we speculate that epithelial dysfunction may only be pathological during the later stages of IPF disease progression where the tissue may no longer be amenable to intervention. Therefore, further investigation into the 1490 genes altered in IPF apices compared to NDC apices may provide important clues into early IPF disease development.
Since the main purpose of this study was to examine the expression of MMP7 in the context of pre-terminal compared to end-stage fibrosis, we used bulk tissue mRNA analyses to maximise the number of independent samples we could investigate. However, using single-cell transcriptomic analyses would have allowed us to specifically identify the transcriptomic profile of cells expressing high levels of MMP7. The NDC donors in our study were significantly younger than the IPF patients, and this is a drawback considering IPF is an age-related disease [37]. Another limitation of our study is that we are not able to determine the activation state of MMP7 produced by the cells we identified as MMP7+. Furthermore, low sample numbers in the multiplex immunohistochemistry analyses prevented investigation of the minority of MMP7+ cells which did not express E-cadherin, CD45, CD31 or αSMA. There are also limitations regarding the range of markers selected for the multiplex analysis. For example, αSMA is not a pan-mesenchymal marker, and therefore we may have missed other fibroblast subsets [38].
Future studies using spatial transcriptomic techniques are warranted to better understand the role of MMP7 in fibrotic tissue remodelling.
The presence of UIP is a marker of poor prognosis no matter the specific ILD diagnosis [39], and elevated MMP7 levels are not specific to IPF [40]. Aberrant MMP7 expression may be a common feature of end-stage fibrotic disease, and mechanisms relating to its regulation and particularly its activation in the fibrotic epithelium should be further investigated.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00191-2022.SUPPLEMENT
Supplementary figures 00191-2022.SUPP_FIGS
Acknowledgements
We thank the patients and their families at the Alfred Hospital for their participation. We would like to thank the Monash Histology Platform for tissue processing. For multiplex immunohistochemistry, we thank the Centre for Advanced Histology and Microscopy (Victorian Comprehensive Cancer Centre). For figures 6a–c and 7 preparation, we thank Yong Kiat Wee (CSL, Ltd)
Footnotes
Provenance: Submitted article, peer reviewed.
Support statement: This study was funded by CSL, Ltd and the National Health and Medical Research Council CRE in Lung Fibrosis, Australia. Funding information for this article has been deposited with the Crossref Funder Registry.
Author contributions: J. Jaffar, L. McMillan and M. Wong conducted experiments, acquired data, and analysed and verified the underlying data. J. Jaffar, M. Wong and G. Westall conceived and designed the study. G.A. Fishbein performed the pathologist's assessment of tissue. M. Alhamdoosh, C. Gamell-Fulla, M. Ng and N. Wilson performed bioinformatic processing and analyses. J. Jaffar, M. Wong, M. Alhamdoosh, C. Gamell-Fulla, I. Glaspole and G. Westall discussed results and their implications. J. Jaffar, G. Westall, K. Symons and I. Glaspole provided patient clinical data and patient tissues. J. Jaffar wrote the manuscript. All authors read and approved the final version of the manuscript.
Conflict of interest: M. Wong has received support for the present manuscript from CSL; the following disclosures have been made outside the submitted work: employee of CSL Innovations Pty Ltd, who provided support for attending meetings/travel; shareholder of CSL stocks. M. Alhamdoosh has received support for the present manuscript from CSL; the following disclosures have been made outside the submitted work: employee of CSL Limited and shareholder of CSL Limited. L. McMillan has received support for the present manuscript from CSL; the following relationships have been disclosed outside the submitted work: employee of CSL Innovations. C. Gamell-Fulla has received support for the present manuscript from CSL. M. Ng reports the following relationships have been disclosed outside the submitted work: employment contract with CSL and stocks owned in CSL. G. Westall has received support for the present manuscript from CSL; the following relationships have been disclosed outside the submitted work: participation in an Advisory Board Meeting for CSL. The remaining authors have nothing to disclose.
- Received April 22, 2022.
- Accepted September 2, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org