





Abstract
Stratification of asthmatic patients based on relevant biomarkers enables the prediction of responsiveness against immune-targeted therapies in patients with asthma. Individualised therapy in patients with eosinophilic asthma has yielded improved clinical outcomes; similar approaches in patients with neutrophilic asthma have yet to be developed. We determined whether colony-stimulating factors (CSFs) in the airway reflect the inflammatory phenotypes of asthma and contribute to disease progression of neutrophilic asthma.
We analysed three different mouse models of asthma and assessed cytokine profiles in sputum from human patients with asthma stratified according to inflammatory phenotype. In addition, we evaluated the therapeutic efficacy of various cytokine blockades in a mouse model of neutrophilic asthma.
Among the CSFs, airway granulocyte CSF (G-CSF) contributes to airway neutrophilia by promoting neutrophil development in bone marrow and thereby distinguishes neutrophilic inflammation from eosinophilic inflammation in mouse models of asthma. G-CSF is produced by concurrent stimulation of the lung epithelium with interleukin (IL)-17A and tumour necrosis factor (TNF)-α; therefore, dual blockade of upstream stimuli using monoclonal antibodies or genetic deficiency of the cytokines in IL-17A×TNF-α double-knockout mice reduced the serum level of G-CSF, leading to alleviation of neutrophilic inflammation in the airway. In humans, the sputum level of G-CSF can be used to stratify patients with asthma with neutrophil-dominated inflammation.
Our results indicated that myelopoiesis-promoting G-CSF and cytokines as the upstream inducing factors are potential diagnostic and therapeutic targets in patients with neutrophilic asthma.
Abstract
Airway G-CSF identifies neutrophil-dominated inflammation in cases of asthma in mice and humans. Dual inhibition of IL-17A and TNF-α effectively ameliorates airway neutrophilia and hyperresponsiveness in a mouse model of neutrophilic asthma. http://bit.ly/36P04dB
Introduction
Most patients with asthma have been successfully managed with standard medications such as inhaled corticosteroids (ICSs) and long-acting β-agonists [1]. However, ∼10% of asthmatic patients show poor treatment adherence to conventional medications [2], which raises the concept of asthma severity and heterogeneity as influencing factors. In terms of inflammation heterogeneity, asthma can be stratified into eosinophilic, neutrophilic, mixed and paucigranulocytic subtypes [3, 4]. Immunological pathways and responsiveness to conventional medications vary between inflammatory phenotypes. Eosinophilic asthma is considered as a type 2 immune response and is relatively well controlled with ICS, while neutrophilic asthma features a type 17 immune response and shows poor response to ICS [5].
Currently, patients showing poor response to ICS are treated with biological inhibitors that target immune molecules such as cytokines and immunoglobulins. However, only a proportion of patients with asthma respond to specific inhibitors and there is a need for patient classification with relevant biomarkers. For patients with eosinophilic asthma, individualised therapy based on biomarkers is well established [6]. Mepolizumab (anti-interleukin (IL)-5) and omalizumab (anti-IgE) were used in the treatment of patients with asthma with high sputum eosinophil counts and IgE levels, respectively, and those patients showed better treatment adherence than patients with low levels of the biomarkers [7, 8]. With regard to treatment of neutrophilic asthma, similar trials focusing on neutrophil or type 17 inflammation [9], including those with anti-IL-17 receptor (IL-17R) [10] and anti-tumour necrosis factor (TNF)-α [11, 12], have been performed. However, an improvement in asthmatic symptoms was not achieved, which may be due to an inability to achieve patient stratification due to lack of adequate markers of neutrophilic asthma.
Colony-stimulating factors (CSFs) consist of three molecular types: macrophage CSF (M-CSF), granulocyte–macrophage CSF (GM-CSF) and granulocyte CSF (G-CSF) [13]. CSFs are considered as regulators of development, survival and function of myeloid cells in the periphery and bone marrow [14]. In particular, inflammation-induced CSFs promote myelopoiesis, which involves the development of monocytes and neutrophils in bone marrow [15, 16]. In terms of asthma, reports have indicated substantial increases in M-CSF and GM-CSF in the airways of patients with eosinophilic asthma [17–19], which facilitates allergen sensitisation and airway eosinophilia in the lung. However, further studies are needed to determine the CSF types that contribute to neutrophilic asthma and their mechanism of action.
In this study, we demonstrated that airway G-CSF distinguishes the inflammatory phenotypes of asthma in mice and humans. In a mouse model of neutrophilic asthma, concurrent stimulation with IL-17A and TNF-α induced G-CSF production in the lung epithelium, which contributed to asthma progression by enhancing neutrophil development in bone marrow, whereas genetic and antibody-mediated depletion of both IL-17A and TNF-α reduced G-CSF production and attenuated airway neutrophilia. In human patients with asthma, sputum analysis revealed that the level of G-CSF is a useful marker for neutrophilic asthma. Based on the results obtained, G-CSF and its associated cytokines are potential diagnostic and therapeutic targets for neutrophilic asthma.
Methods
Detailed methods are provided in the supplementary material.
Study approval and subjects
Animal experiments were performed in accordance with animal guidelines of the Animal Research Committee of Pohang University of Science and Technology (POSTECH), Gyeongbuk, Republic of Korea (POSTECH-2017-0109-C1). Informed written consent was obtained from all participants. The protocol was approved by the Ethics Committee of Soonchunhyang University Bucheon Hospital, Bucheon, Republic of Korea (SCHBC-2017-07-017-002). The diagnosis of asthma was based on Global Initiative for Asthma guidelines [1]. Patients with parenchymal lung disease were excluded. Patients were categorised according to the inflammatory subtype of their sputum sample [3].
Mouse models of asthma
Mice aged 6–8 weeks old were used. For the lipopolysaccharide (LPS)/ovalbumin (OVA)-induced model, mice were intranasally sensitised with 10 µg LPS plus 75 µg OVA. For the aluminium hydroxide (alum)/OVA-induced model, mice were intraperitoneally sensitised with 2 mg alum plus 75 µg OVA. During challenges, 50 µg OVA in 20 µL PBS was intranasally introduced on 2 consecutive days per week for 2 or 4 weeks starting on day 14 for the alum/OVA or LPS/OVA models, respectively. For the house dust mite (HDM)-induced model, mice were intranasally sensitised and challenged with 25 µg HDM (Greer Laboratories, Lenoir, NC, USA) on days 0, 1, 2, 14, 15, 16 and 17.
Statistics
Statistical analysis was performed using Prism (GraphPad, La Jolla, CA, USA) and SPSS Statistics (IBM, Armonk, NY, USA). Data are presented as mean±sem or median as indicated. p-values <0.05 were considered to be significant.
Results
Airway G-CSF identifies neutrophil-dominated inflammation in mouse models of asthma
To assess the impact of CSFs on different inflammatory phenotypes of asthma, three mouse models of asthma were established: HDM-, alum/OVA- and LPS/OVA-induced asthma [20–22]. In the alum/OVA-induced asthma model, an increase in both the airway neutrophil and eosinophil counts was observed (figure 1a), whereas in the LPS/OVA- and HDM-induced models, specific increases in neutrophil and eosinophil counts, respectively, were observed (figure 1b and c). Next, in the results of the evaluation of airway CSFs, M-CSF was undetectable in all groups (figure 1d), while GM-CSF was slightly increased only in the alum/OVA model (figure 1e). Interestingly, G-CSF was significantly increased in the LPS/OVA- and alum/OVA-induced models in order, but not in the HDM-induced model (figure 1f). In the association analysis, positive correlations between G-CSF levels and neutrophil frequency (figure 1g) and inverse correlations between G-CSF levels and eosinophil frequency (figure 1h) were obtained. The phenotypes of airway inflammation among different animal models of asthma were determined by the types of adjuvants used to induce immune responses rather than the different time courses of each animal model. Synchronising the period and number of allergen challenges in various asthma models also elicited distinct inflammatory patterns in the airway (supplementary figure S1a–f). These data demonstrate that the airway G-CSF level identifies neutrophil-dominated inflammation in mouse models of asthma.

Airway granulocyte colony-stimulating factor (G-CSF) level distinguishes neutrophilic inflammation from eosinophilic inflammation in mouse models of asthma. i.p.: intraperitoneally; OVA: ovalbumin; BALF: bronchoalveolar lavage fluid; i.n.: intranasally; LPS: lipopolysaccharide; HDM: house dust mite; M-CSF: macrophage CSF; GM-CSF: granulocyte–macrophage CSF. a–c) Experimental schemes of mouse models of asthma. Total and immune cell counts in BALF. n=7–11 for each group. a) Alum/OVA-induced mixed-type asthma. b) LPS/OVA-induced neutrophilic asthma. c) HDM-induced eosinophilic asthma. d–f) Concentration of CSFs in BALF: d) M-CSF, e) GM-CSF and f) G-CSF. g, h) Correlations between BALF G-CSF with airway g) neutrophil and h) eosinophil frequency. Two or three independent experiments were performed and represented as median for (a)–(f). p-values were determined by the Mann–Whitney U-test for (a)–(c), the Kruskal–Wallis test with post hoc analysis for (d)–(f) and the nonparametric Spearman's correlation test for (g) and (h). **: p<0.01; ***: p<0.001.
G-CSF contributes to airway neutrophilia through upregulated myelopoiesis in bone marrow
The functional roles of G-CSF in airway neutrophilia were evaluated using the LPS/OVA-induced mouse model. In response to G-CSF neutralisation, neutrophil infiltration in bronchoalveolar lavage fluid (BALF) and the lung (figure 2a–c) was attenuated and inflammatory scores in the lung (figure 2d) were reduced, which indicated that inflammation-induced G-CSF was associated with airway neutrophilia. Next, the role of G-CSF in survival, recruitment and activation of neutrophils in the airway was assessed [23]. The proportion of early apoptotic neutrophils (7-aminoactinomycin D (7-AAD)−Annexin V+) was decreased in mice with inflammation regardless of G-CSF inhibition compared with control mice, while the proportion of late apoptotic neutrophils (7-AAD+Annexin V+) showed no significant difference among groups (figure 2e). In addition, the expression levels of CD62L (figure 2f) and myeloperoxidase (figure 2g), which reflect neutrophil activation, and that of the neutrophil-recruiting chemokine CXCL1 (figure 2h) were not attenuated by G-CSF blockade.
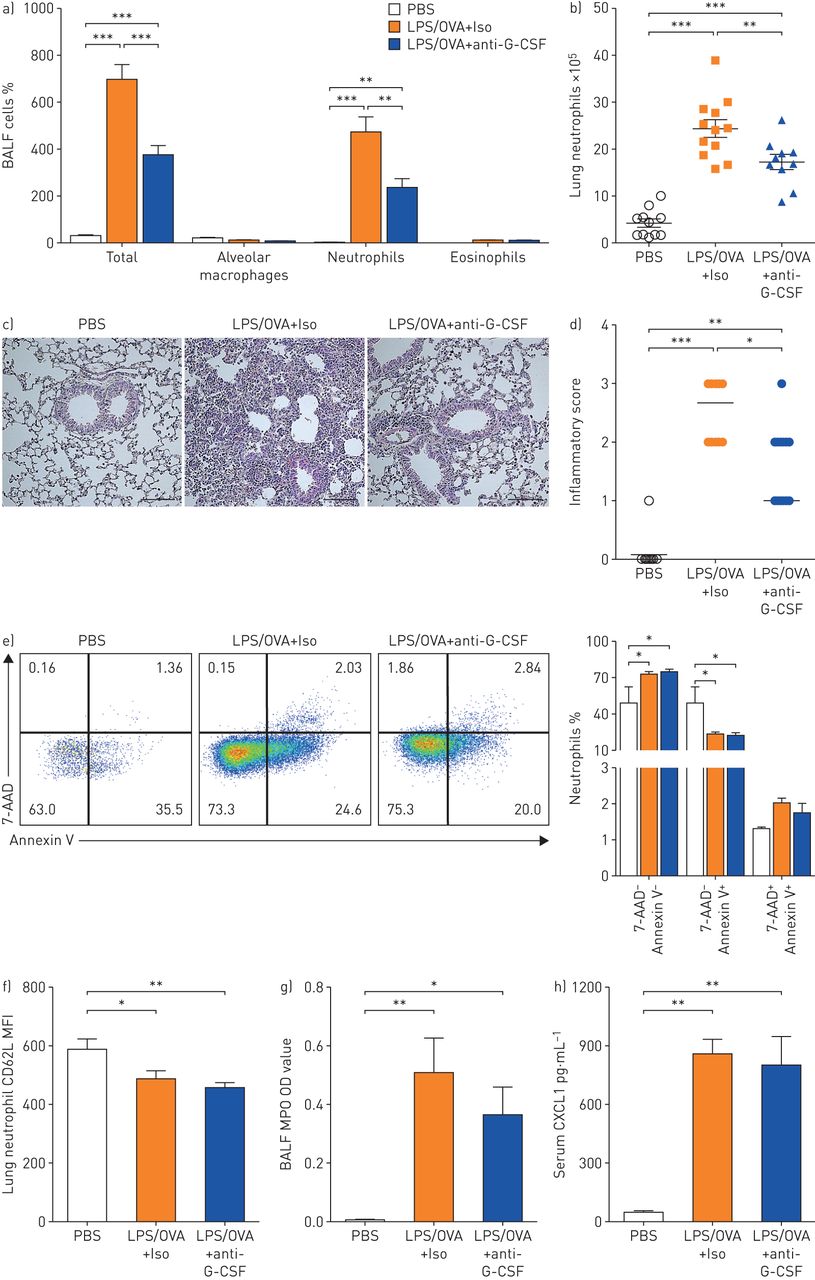
Granulocyte colony-stimulating factor (G-CSF) facilitates airway neutrophilia irrespective of in situ regulation at the lung. BALF: bronchoalveolar lavage fluid; LPS: lipopolysaccharide; OVA: ovalbumin; Iso: isotype; 7-AAD: 7-aminoactinomycin D; MFI: mean fluorescent intensity; MPO: myeloperoxidase; OD: optical density. a–h) G-CSF was ablated by anti-G-CSF in an LPS/OVA-induced model. a) Total and immune cell counts in BALF. b) Neutrophil numbers in lung tissues. n=11–12 for each group. c) Representative haematoxylin/eosin-stained lung tissues. Scale bar: 100 µm. d) Perivascular and peribronchial inflammation. e) Neutrophil apoptosis was evaluated by Annexin V and 7-AAD expression: representative flow cytometry plots and bar graphs of early and late apoptotic neutrophils. CD11b+Gr-1+ gated. f) MFI of CD62L on lung neutrophils. g) MPO activity in BALF. h) Concentration of CXCL1 in serum. n=10–11 for each group. Two or three independent experiments were performed and represented as mean±sem for (a), (b) and (e)–(h) and median for (d). p-values were determined by one-way ANOVA with Bonferroni's multiple comparison test for (a), (b) and (e)–(h) and the Kruskal–Wallis test with post hoc analysis for (d). *: p<0.05; **: p<0.01; ***: p<0.001.
In the results of the cytokine analysis, G-CSF was a predominant cytokine in serum of mice with airway neutrophilia, while the other inflammatory cytokines were mostly below detectable levels (figure 3a and b). Since circulating G-CSF functions as a haematopoietic regulator [23], changes of haematopoiesis in bone marrow were analysed. Lineage−Sca-1+c-Kit+ (LSK) cells, which comprise both haematopoietic stem and progenitor cells, were significantly higher in mice with airway neutrophilia than in control mice (figure 3c). In addition, granulocyte and monocyte progenitor (GMP) cells (figure 3c) and CD11b+ myeloid lineage cells containing neutrophils were upregulated, while B220+ B-lymphocytes were downregulated (figure 3d). In agreement with the results obtained using the LPS/OVA-induced model, upregulation of myelopoiesis was observed in mice that were administered recombinant murine (rm) G-CSF (figure 3e and f). Moreover, the inflammation-induced dysregulation of haematopoiesis in bone marrow was improved by G-CSF blockade; downregulation of the LSK cells and myeloid lineage cells was observed, although GMP cells were not changed (figure 3g and h). Collectively, the results suggest that G-CSF may contribute to airway neutrophilia through its haematopoietic regulatory function as a mediator of physiological cross-talk between the lung and bone marrow, rather than through its role as an in situ immune modulator of neutrophils in the lung.

Granulocyte colony-stimulating factor (G-CSF) promotes airway neutrophilia by acting as a long-range haematopoietic factor enhancing myelopoiesis in bone marrow (BM). IL: interleukin; M-CSF: macrophage CSF; GM-CSF: granulocyte–macrophage CSF; TNF: tumour necrosis factor; IFN: interferon; OVA: ovalbumin; LSK: lineage−Sca-1+c-Kit+; MP: myeloid progenitor; CMP: common MP; GMP: granulocyte and monocyte progenitor; MEP: megakaryocyte and erythrocyte progenitor; LPS: lipopolysaccharide; rm: recombinant murine. a) Heatmap of cytokines in serum from control and inflamed mice at 6, 12, 24 and 48 h after the last OVA challenge (n=5–6 for each group, other cytokines include M-CSF, GM-CSF, IL-1β, TNF-α, IL-4, IL-17A and IFN-γ). Each row indicates the concentration of cytokines from individual mice. Each column indicates each time-point. b) Kinetics of G-CSF concentration in serum of control and inflamed mice. n=5–6 for each group. c, d) Frequency of LSK, committed progenitor and lineage cells in BM after disease induction. n=11–12 for each group. e, f) Frequency of LSK, committed progenitor and lineage cells in BM after rmG-CSF injection. n=7–8 for each group. g, h) Frequency of LSK, committed progenitor and lineage cells in BM from PBS-, LPS/OVA+isotype- and LPS/OVA+anti-G-CSF-treated mice. n=10–13 for each group. Two or three independent experiments were performed and represented as median for (b), (e) and (f) and mean±sem for (c), (d), (g) and (h). p-values were determined by the Kruskal–Wallis test with post hoc analysis for (b), the unpaired two-tailed t-test for (c) and (d), the Mann–Whitney U-test for (e) and (f) and one-way ANOVA with Bonferroni's multiple comparison test for (g) and (h). *: p<0.05; **: p<0.01; ***: p<0.001.
Lung epithelial cells produce G-CSF in response to concurrent stimulation with IL-17A and TNF-α
The cellular sources of G-CSF and associated inducing factors during airway neutrophilia were determined. Under consideration that G-CSF is produced by haematopoietic and nonhaematopoietic cells [24], CD45+ haematopoietic cells, CD45−EpCAM+ epithelial cells and CD45−CD31+ endothelial cells were isolated from inflamed lung tissues, and G-CSF expression was analysed. EpCAM+ lung epithelial cells composed of the alveolar and bronchial epithelial compartments [25] showed higher expression of G-CSF mRNA than haematopoietic and endothelial cells during airway neutrophilia (figure 4a). IL-17A and TNF-α cooperatively induced G-CSF from various nonhaematopoietic cells in vitro [26, 27]. Since those cytokines were increased in the inflamed lung in the LPS/OVA model (supplementary figure S2a), the mouse and human lung epithelial cell lines were stimulated with rmIL-17A and rmTNF-α, either singly or in combination. In both mouse and human lung epithelial cell lines, G-CSF production was higher under combined treatment of rmIL-17A and rmTNF-α versus single treatments (supplementary figure S2b–d), which is consistent with the findings of a previous study [28].
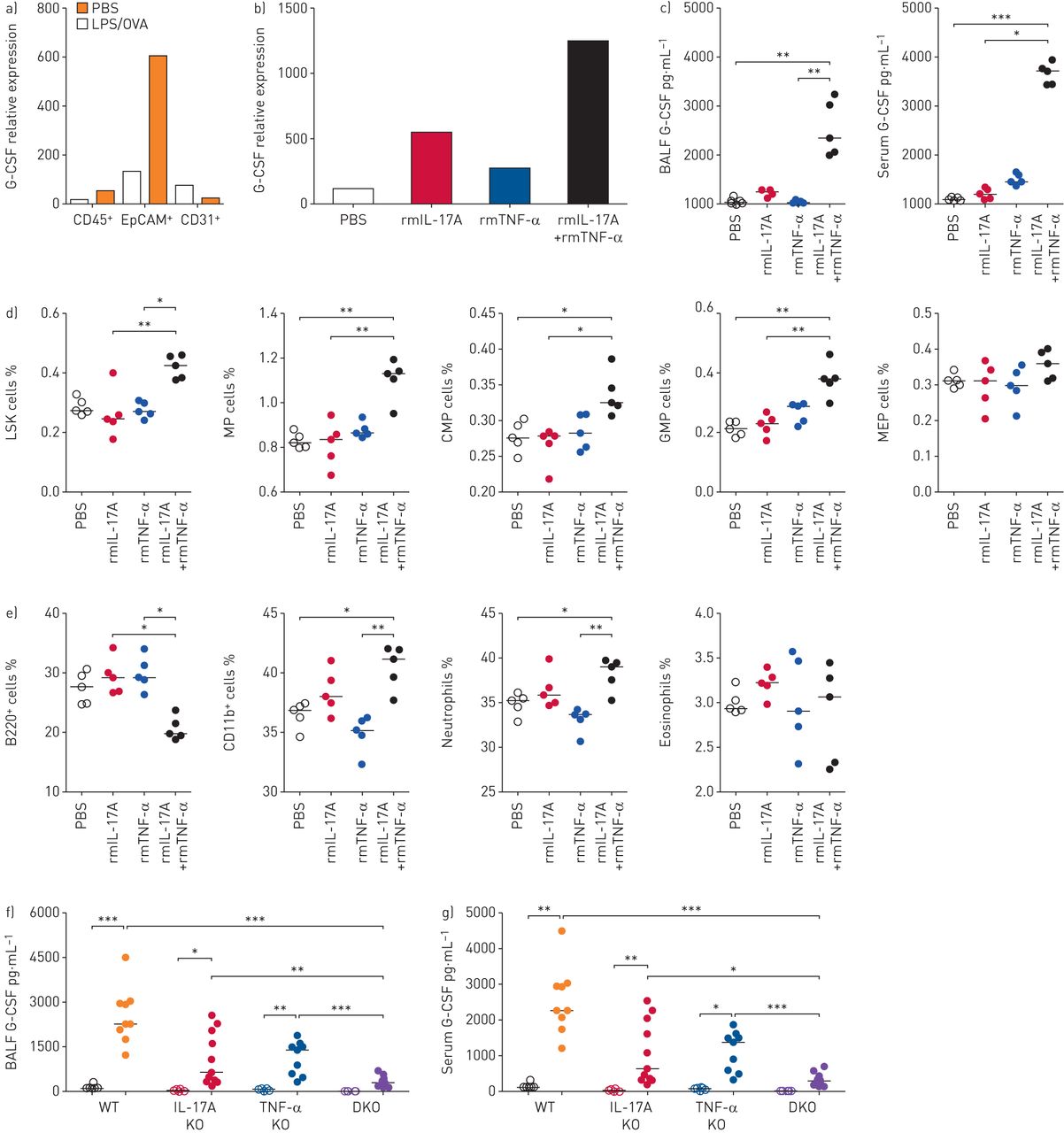
Lung epithelial cells produce granulocyte colony-stimulating factor (G-CSF) in response to concurrent stimulation of interleukin (IL)-17A and tumour necrosis factor (TNF)-α. LPS: lipopolysaccharide; OVA: ovalbumin; BALF: bronchoalveolar lavage fluid; rm: recombinant murine; BM: bone marrow; LSK: lineage−Sca-1+c-Kit+; MP: myeloid progenitor; CMP: common MP; GMP: granulocyte and monocyte progenitor; MEP: megakaryocyte and erythrocyte progenitor; WT: wild-type; KO: knockout; DKO: double-KO. a) mRNA levels of G-CSF in FACS-sorted CD45+ haematopoietic, EpCAM+ lung epithelial and CD31+ endothelial cells from PBS- or LPS/OVA-treated mice. n=4–5 mice were pooled by group for cell sorting. b) mRNA level of G-CSF in FACS-sorted EpCAM+ lung epithelial cells. n=5 mice were pooled by group for cell sorting. c) Protein levels of G-CSF in BALF and serum. n=5 in all groups. d, e) Frequency of LSK, committed progenitor and lineage cells in BM of PBS- or cytokine-treated groups. n=5 in all groups. f, g) Protein levels of G-CSF in f) BALF and g) serum of WT, IL-17A KO, TNF-α KO and DKO mice after PBS or LPS/OVA treatment. n=6–11 for each group. Empty and filled circles indicate PBS and LPS/OVA groups, respectively. Two or three independent experiments were performed and represented as median for (c)–(g). p-values were determined by the Kruskal–Wallis test with post hoc analysis for (c)–(g). *: p<0.05; **: p<0.01; ***: p<0.001.
The physiological roles of IL-17A and TNF-α in G-CSF production and airway neutrophilia in vivo were assessed using two complementary experimental approaches in mice. For a gain-of-function study, rmIL-17A and rmTNF-α were administered intranasally in mice, and G-CSF levels were subsequently measured. The expression level of G-CSF mRNA in lung epithelial cells (figure 4b) and the protein levels of G-CSF in BALF and blood (figure 4c) were robustly enhanced by co-administration of the two cytokines. Consistent with the results using the LPS/OVA model, enhanced myelopoiesis in bone marrow (figure 4d and e) and increased pulmonary neutrophil infiltration (supplementary figure S3a and b) and CXCL1 in the circulation (supplementary figure S3c) were observed in the mice with IL-17A and TNF-α co-treatment. For a loss-of-function study, the LPS/OVA-induced model was established in wild-type (WT), IL-17A knockout (KO), TNF-α KO and IL-17A×TNF-α double-KO (DKO) mice. Greater reduction in the level of G-CSF was observed in DKO mice than in single-KO mice, which indicates that both cytokines are required for G-CSF induction during neutrophilic inflammation (figure 4f and g). Moreover, airway neutrophilia and CXCL1 production were decreased to lower levels in DKO mice than in WT or single-KO mice (supplementary figure S3d–f). These results indicate that both IL-17A and TNF-α are upstream factors for inducing G-CSF and neutrophil recruitment in the airway.
Simultaneous blockade of IL-17A and TNF-α alleviates airway neutrophilia and airway hyperresponsiveness
As a therapeutic approach for neutrophilic asthma, concurrent blockade of both IL-17A and TNF-α using neutralising antibodies was carried out during the disease course (figure 5a). In the mice with dual cytokine blockade, substantial decreases of G-CSF levels were observed in BALF and serum (figure 5b), and dysregulated haematopoiesis in bone marrow was improved (figure 5c and d). Consistent with the bone marrow phenotype, neutrophil infiltration in BALF and the lung (figure 5e–g) and the inflammatory scores in the lung (figure 5h) were reduced by treatments. The addition of exogenous rmG-CSF in IL-17A/TNF-α-depleted mice restored enhanced myelopoiesis in bone marrow and neutrophilic inflammation in the lung, suggesting a direct role of G-CSF as a haematopoietic regulator during neutrophilic asthma (supplementary figure S4a–d). The production of CXCL1 was significantly decreased under dual blockade (figure 5i), which was not shown during G-CSF blockade. Development of airway hyperresponsiveness is a conspicuous symptom of asthma [29] and in our study the symptom was decreased in mice with dual blockade (figure 5j). With regard to the therapeutic efficacy of cytokine blockade, significantly better performance was observed for dual blockade versus single blockade (supplementary figure S5a–h), which indicates that dual blockade of IL-17A and TNF-α has potential as a therapeutic intervention in patients with neutrophilic asthma through its effectiveness to attenuate haematopoietic disturbance in bone marrow and reduce neutrophil recruitment in the airway.

Simultaneous ablation of granulocyte colony-stimulating factor (G-CSF)-inducing factors, interleukin (IL)-17A and tumour necrosis factor (TNF)-α, significantly ameliorates the inflammation in bone marrow (BM) and airways. i.n.: intranasally; OVA: ovalbumin; LPS: lipopolysaccharide; i.p.: intraperitoneally; Iso: isotype; BALF: bronchoalveolar lavage fluid; LSK: lineage−Sca-1+c-Kit+; MP: myeloid progenitor; CMP: common MP; GMP: granulocyte and monocyte progenitor; MEP: megakaryocyte and erythrocyte progenitor; Rrs: resistance of the respiratory system. a) Experimental scheme of blocking IL-17A and TNF-α. b) Levels of G-CSF in BALF and serum. n=10–11 for each group. c, d) Frequency of LSK, committed progenitor and lineage cells in BM from PBS-, LPS/OVA+Iso- and LPS/OVA+anti-IL-17A+anti-TNF-α-treated mice. n=10–12 for each group. e) Total and immune cell counts in BALF. f) Neutrophil numbers in lung tissues. n=10–11 for each group. g) Representative haematoxylin/eosin-stained lung tissues from PBS, LPS/OVA+Iso and LPS/OVA+anti-IL-17A+anti-TNF-α mice. Scale bar: 100 µm. h) Perivascular and peribronchial inflammation. i) Concentration of CXCL1 in serum. j) Airway hyperresponsiveness (assessed by changes in Rrs). n=6–10 for each group. Two or three independent experiments were performed and represented as mean±sem for (b)–(f) and (j) and median for (i) and (h). p-values were determined by one-way ANOVA with Bonferroni's multiple comparison for (b)–(f), the Kruskal–Wallis test with post hoc analysis for (i) and (h) and two-way ANOVA with repeated measurement for (j). *: p<0.05; **: p<0.01; ***: p<0.001.
Sputum levels of G-CSF and TNF-α stratify neutrophil-dominated inflammation in human patients with asthma
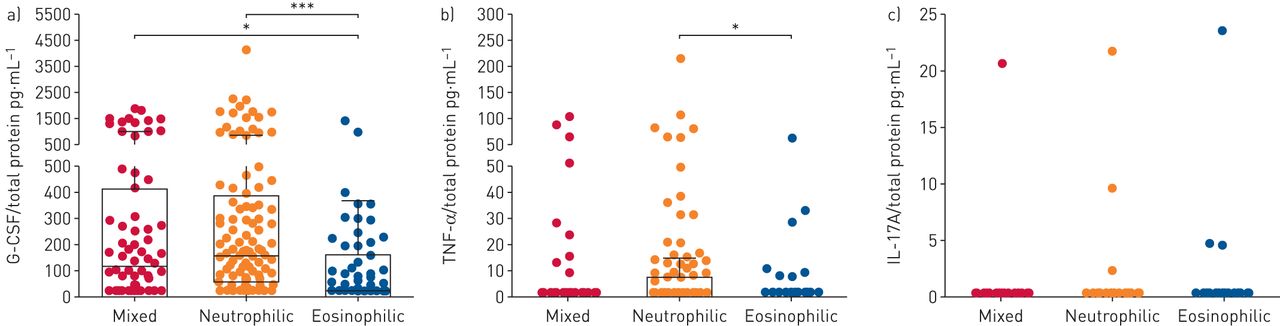
Considering the findings in the mice models, levels of G-CSF and upstream inducer cytokines in sputum were evaluated in human patients with asthma stratified by inflammatory cell infiltrates as mixed, neutrophilic or eosinophilic type (supplementary table S1) [30]. The sputum G-CSF level was significantly higher in patients with neutrophilic asthma than in those with eosinophilic asthma (figure 6a) and the TNF-α level discriminated neutrophilic asthma from eosinophilic asthma (figure 6b). Among the cytokines, G-CSF was detected in many patients, while TNF-α and IL-17A were detected at very low levels or were undetectable (figure 6b and c), indicating that G-CSF exhibits greater sensitivity than other cytokines by ELISA-based quantification. Collectively, the results obtained from clinical samples reveal that the levels of sputum G-CSF and upstream cytokines have potential as diagnostic and therapeutic targets in patients with neutrophilic asthma.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sputum granulocyte colony-stimulating factor (G-CSF) identifies neutrophil-dominated asthma in humans. TNF: tumour necrosis factor; IL: interleukin. a–c) Concentration of sputum a) G-CSF, b) TNF-α and c) IL-17A in cellularity-based subcategorised asthmatic patients. Concentrations of cytokines were normalised to total protein amount. Box and whisker plots show median and interquartile range with upper and lower inner fences (Tukey). p-values were determined by the Kruskal–Wallis test and post hoc analysis. *: p<0.05; ***: p<0.001.
Discussion
We used a mouse model of neutrophilic asthma to delineate the mechanism of action of G-CSF to contribute to neutrophilic inflammation (supplementary figure S6). Furthermore, by assessing cytokines in sputum of asthmatic patients, we demonstrated the usefulness of the levels of G-CSF and associated cytokines as diagnostic and therapeutic targets for neutrophilic asthma. Our results indicated: 1) a substantial increase of the airway G-CSF in the mouse model of neutrophilic asthma and human asthmatic patients with neutrophil-dominated inflammation; 2) G-CSF production of the lung epithelial cells with concurrent stimulation of IL-17A and TNF-α; 3) G-CSF-mediated airway neutrophilia through long-range haematopoietic regulation of increases in neutrophil generation; 4) alleviation of increased G-CSF production and airway neutrophilia in a genetic deletion model of IL-17A and TNF-α DKO mice; and 5) effective performance of dual blockade of IL-17A and TNF-α with monoclonal antibodies to achieve therapeutic efficacy in the case of neutrophilic asthma.
CSFs were first defined as haematopoietic growth factors that generate colonies of myeloid cells in vitro [31]. Subsequently, studies revealed that CSFs are involved in the pathogenesis of inflammatory diseases by promoting the development, survival and functions of myeloid cells [13]. With regard to asthma, some recent studies revealed that M-CSF and GM-CSF are involved in allergen sensitisation and eosinophil accumulation in the airway [17, 18]. In contrast to the other CSFs, G-CSF negatively correlates with T-helper (Th) type 2 immune responses in patients with eosinophilic asthma [32] and exogenous G-CSF treatment suppresses airway eosinophilia in a mouse model [33], suggesting distinct immune-modulatory roles of G-CSF in asthma. To the best of our knowledge, the current study is the first to indicate that G-CSF is significantly associated with neutrophilia but not with eosinophilia in the airways in mouse models of asthma and human patients with neutrophilic asthma. G-CSF production has been reported in various inflammatory lung diseases such as pneumonia [34], bronchiectasis [35] and chronic obstructive pulmonary disease (COPD) [36]; nevertheless, we considered that the increased levels of G-CSF in the airways of the mouse models of asthma and human asthmatic patients found in this study are unique phenomena of asthma. In the LPS/OVA-induced mouse model, G-CSF is elicited by IL-17 produced by immune cells such as Th17 cells, type 3 innate lymphoid cells and γδ T-cells rather than infections and endotoxin infusion which also cause G-CSF production and nonasthmatic neutrophilic airway inflammation in mice [16]. Moreover, we excluded patients with parenchymal lung diseases including bronchiectasis, pulmonary tuberculosis and COPD from the human asthma cohort of our study. Collectively, our results indicated that the airway G-CSF delineates neutrophilic asthma.
The lung epithelial cells are exposed to various inflammatory stimuli and, in turn, generate appropriate responses to resolve inflammation [37, 38]. A recent study reported that stimuli and derived mediators of the lung epithelium were potent therapeutic targets and predictive biomarkers for asthma [8]. In our study including a mouse model of neutrophilic asthma, we demonstrated the pathophysiological role of the lung epithelial responses induced by concurrent stimulation with IL-17A and TNF-α; the two modes of epithelial responses in neutrophilic asthma include in situ local modulations at the lung via neutrophil-recruiting chemokines, including CXCL1 [39], and long-range regulation of haematopoiesis at the bone marrow via G-CSF. The findings of the current study indicate that simultaneous blockade of the divergent epithelial responses is effective to mitigate disease progression of neutrophilic asthma. Indeed, blockade of G-CSF alone was not sufficient to reduce overall airway neutrophilia, due to lack of effect of the modality on CXCL1 production. In contrast, dual blockade of upstream IL-17A and TNF-α by using monoclonal antibodies or genetic deficiency of those cytokines in DKO mice dramatically ameliorated inflammation by reducing CXCL1 and G-CSF production.
Research and development for effective therapy in patients with severe asthma is considered to be important. Since asthma has heterogeneous inflammatory attributes, relevant biomarkers for stratification of asthmatic patients would enable the prediction and monitoring of responses to targeted therapeutics, which is a desirable goal [8]. However, no validated biomarkers for neutrophilic asthma are currently available. We considered that Th17 mediators, such as IL-17A and TNF-α, are potential targets to treat neutrophilic asthma in patients with appropriate stratified type. Recent clinical trials focused on IL-17R [10] or TNF-α [11, 12] have provided valuable insights; single-cytokine blockade in nonstratified severe asthmatic patients failed to achieve any significant improvement. Nevertheless, reports have indicated clinical responses to therapies in some subgroups of patients: patients with high reversibility responded well to anti-IL-17R [10], aged patients showed a trend towards lower risk of exacerbation in response to the monoclonal TNF-α antibody golimumab [11] and some patients with refractory asthma showed improvement in asthma-related quality-of-life scores in response to the soluble TNF-α receptor etanercept [12]. These data collectively suggest that appropriate specification of asthmatic patients may be a crucial factor for determining the success of targeted therapies.
Our study demonstrated that dual blockade of IL-17A and TNF-α was effective against neutrophilic inflammation, while single-cytokine blockade of either IL-17A or TNF-α achieved less therapeutic benefits in our model, which suggests that synergistic stimulation of the lung epithelium by the two cytokines is a key mechanism. This translational study highlights the clinical applicability of targeting the lung epithelium in patients with asthma based on the following findings: 1) G-CSF is produced by human lung epithelial cell lines in response to IL-17A and TNF-α stimulation, and 2) the sputum level of G-CSF differentiates between patients with neutrophil-dominant asthma and those with eosinophil-dominant asthma. Collectively, these results could enable stratification of patients with asthma according to the level of airway G-CSF and inflammatory cell type in sputum. The proposed stratification strategy has potential use as a new therapeutic approach in human patients with neutrophilic asthma by targeting the lung epithelium responses with simultaneous blockade of IL-17A and TNF-α.
Our study has some limitations that require further investigation. First, although we designated the pre-clinical models of asthma in mice, they do not fully represent the pathology of asthma in humans, and investigators should test the association of the airway G-CSF and airway neutrophilia in other models. Second, although we reported that the lung epithelium is a source of G-CSF based on the results of analysis of G-CSF mRNA, further studies focused on the subsets of lung epithelial cells that produce G-CSF protein are needed. Finally, investigation on the mode of action of G-CSF as a haematopoietic regulator in human asthmatic patients is required.
In summary, the lung epithelial responses of G-CSF-associated pathway activation had a role in the pathophysiology of neutrophilic asthma. Our study will enable the development of novel treatments for other inflammatory diseases involving excessive neutrophils, such as COPD, psoriasis and inflammatory bowel disease.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00827-2019.SUPPLEMENT
Supplementary table and figures ERJ-00827-2019.FIGURES
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00827-2019.Shareable
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: Y-M. Kim designed the study, did experiments, analysed data and wrote the manuscript; H. Kim, S. Kim, H.W. Park and G. You helped with the mouse experiments and edited the manuscript; Seungwon Lee made initial observations by analysing mouse samples; H. Kang and Seyoung Lee helped with in vitro experiments; J-U. Lee performed human statistical analyses; H-S. Park and Y. Choi helped with the experiments measuring airway hyperresponsiveness; Y. Park edited the manuscript; C-S. Park and J-S. Park provided sputum samples of asthmatic patients; S-W. Lee was the principal investigator for all human and mouse studies, and supervised the project.
Conflict of interest: Y-M. Kim has a patent 10-2019-0057637 pending.
Conflict of interest: H. Kim has nothing to disclose.
Conflict of interest: Seungwon Lee has nothing to disclose.
Conflict of interest: S. Kim has a patent 10-2019-0057637 pending.
Conflict of interest: J-U. Lee has nothing to disclose.
Conflict of interest: Y. Choi has nothing to disclose.
Conflict of interest: H.W. Park has nothing to disclose.
Conflict of interest: G. You has nothing to disclose.
Conflict of interest: H. Kang has nothing to disclose.
Conflict of interest: Seyoung Lee has nothing to disclose.
Conflict of interest: J-S. Park has nothing to disclose.
Conflict of interest: Y. Park has nothing to disclose.
Conflict of interest: H-S. Park has nothing to disclose.
Conflict of interest: C-S. Park has a patent 10-2019-0057637 pending.
Conflict of interest: S-W. Lee has a patent 10-2019-0057637 pending.
Support statement: This study was supported by BK21 Plus program and a grant funded by the Ministry of Education of Korea (10Z20130012243; 2018R1A6A3A01011004), grants funded by the National Research Foundation of Korea (NRF-2017R1A5A1015366; NRF-2017R1A2B4005400; NRF-2017R1A2B4012693), and a grant from the Korean Health Technology R & D Project by the Ministry of Health and Welfare of Korea (HI16C0992). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 26, 2019.
- Accepted November 5, 2019.
- Copyright ©ERS 2020
References










