





Abstract
This review provides a pulmonary-focused description of the age-associated changes in the integrative physiology of exercise, including how declining lung function plays a role in promoting multimorbidity in the elderly through limitation of physical function. We outline the ageing of physiological systems supporting endurance activity: 1) coupling of muscle metabolism to mechanical power output; 2) gas transport between muscle capillary and mitochondria; 3) matching of muscle blood flow to its requirement; 4) oxygen and carbon dioxide carrying capacity of the blood; 5) cardiac output; 6) pulmonary vascular function; 7) pulmonary oxygen transport; 8) control of ventilation; and 9) pulmonary mechanics and respiratory muscle function. Deterioration in function occurs in many of these systems in healthy ageing. Between the ages of 25 and 80 years pulmonary function and aerobic capacity each decline by ∼40%. While the predominant factor limiting exercise in the elderly likely resides within the function of the muscles of ambulation, muscle function is (at least partially) rescued by exercise training. The age-associated decline in pulmonary function, however, is not recovered by training. Thus, loss in pulmonary function may lead to ventilatory limitation in exercise in the active elderly, limiting the ability to accrue the health benefits of physical activity into senescence.
Abstract
Exercise performance depends on integrated organ-system response, each subject to differential age-related decline http://ow.ly/YRNh3022S7w
Introduction
Human ageing is a condition satisfying four principles: it is intrinsic, universal, progressive and usually detrimental to the host [1]. The proportion of the world's population over the age of 60 years increased from 9.2% in 1990 to 11.7% in 2013 and is projected to be 21.1%, or 2 billion people, by 2050 [2]. Ageing is associated with loss of physical function. The complex interplay between age-associated reduction in habitual physical activity and the intrinsic ageing processes complicates interpretation of the aetiology of physical function decline. Nevertheless, that physical inactivity is a primary cause of most chronic diseases [3] means that ability to maintain physical function into older age is vital to extend the time lived in optimal health: the “healthspan” [4]. Increasing older adults' healthspan could dramatically lessen the individual and societal impact of an ageing population. Indeed, prevalence of multimorbidity (two or more long-term disorders) is much greater in the elderly: present in 65% of individuals aged 65–84 years, and 82% of people above 85 years old [5]. This review will explore contributors to exercise limitation in senescence, with a special focus on the lung.
Muscular exercise poses a systemic stress to homeostasis that demands an integrated multi-organ response. While physical activity was a key evolutionary stressor that contributed to shaping structure and function of human organ systems, prevalence of both chronic inactivity and increasing longevity poses a new challenge for the modern human to meet systemic demands of exercise into old age. Poor performance on cycle, treadmill or endurance walking tests in old age indicates proximity to future health decline [6]. This suggests a fundamental connection between aerobic capacity (maximal oxygen uptake (V′O2max)) and longevity. Animal studies of artificial selective breeding for running capacity show that high V′O2max is associated with an ∼25% survival increase, lower mean arterial pressure, circulating cholesterol and triglycerides, and increased glucose tolerance, among many other health-associated effects [7]. Interestingly, while the lung is often touted as “overbuilt” for exercise, selective breeding for aerobic capacity hints otherwise. Allometrically scaled lung volume is greater in rats bred for high V′O2max while, at maximal exercise, alveolar ventilation and effective pulmonary diffusing capacity are greater, and arterial CO2 partial pressure (PaCO2) and pulmonary vascular resistance are less than in rats bred for low V′O2max [8]. These findings support case reports in humans that supra-normal pulmonary function is required to allow adequate breathing reserve for youthful V′O2max maintenance into old age [9].
V′O2max declines with age (figure 1a) [12–15]. This is likely related, in part, to physical inactivity co-incident with advancing age: octogenarian endurance athletes can maintain V′O2max close to the median of those 40 years younger (38 mL·min−1 kg−1) [16] and in some cases younger still (50 mL·min−1·kg−1) [9]. Nevertheless, cross-sectional studies suggest that V′O2max declines with a rate between 0.2 and 0.5·mL·min−1·kg−1 year−1 (∼0.5% per year) after the age of 30 years, while longitudinal studies suggest that V′O2max decline may accelerate after ages 40–50 years [12, 17]. The 810 healthy men and women studied in the Baltimore Longitudinal Study of Aging between 1978 and 1998 (median follow-up 7.9 years) revealed that V′O2max decline accelerated from ∼0.3 to 0.6% per year in the 20–30-year-old group to >2% per year in 70-79-year-age group and beyond, even when scaled to fat free mass [12]. Aetiology of accelerated loss in older age is multifactorial, and may be consequent to greater rate of decline in stroke volume and muscle O2 extraction after 50 years of age [17–20], compared with relatively linear, and smaller magnitude, decline in maximal heart rate [12]. Despite accelerated V′O2max decline in older age, greater habitual physical activity at any age is accompanied by greater V′O2max [12, 16]. Even in octogenarians, habitual endurance exercise is associated with greater muscle oxidative capacity and expression of transcription factors associated with mitochondrial biogenesis [16]. Thus, maintaining physical activity in older age is associated with greater central (cardiac output) and peripheral (muscle O2 extraction) capacity compared with sedentary senescence.

a) Decline in maximal oxygen uptake (V′O2max) with age. V′O2max is expressed as a % predicted value [10] of a 25-year-old individual of average weight and height (male 177 cm height and 82 kg weight; female 164 cm height and 65 kg weight). b) Decline in forced expiratory volume in 1 s (FEV1) with age. FEV1 is expressed as a % predicted value [11] for a 25-year-old individual of the same weight and height as in (a).
The lactate threshold also declines with age [21, 22]. Cross-sectional studies suggest that lactate threshold decline (both in absolute terms and relative to mass) is less rapid than V′O2max, such that lactate threshold/V′O2max in untrained subjects increases from ∼40–50% in youth to ∼55–70% in the 70-79-year-age group; an effect that may be more pronounced in women than men.
Pulmonary function, however, does not respond to exercise training [23]. Therefore, age-related decline in pulmonary function (figure 1b) may become an increasingly important limiting factor for physical activity and V′O2max in the elderly. Inevitable loss in lung elastic recoil associated with ageing leads to pulmonary mechanics alterations and a tendency to ventilatory limitation in older individuals. In most elderly subjects, physical activity decline may be considered protective against development of exertional symptoms and exercise limitation. However, typical lifelong pulmonary function and V′O2max declines are roughly proportional (figure 1), meaning that ventilatory limitation may become more noticeable in elderly who maintain high levels of physical activity.
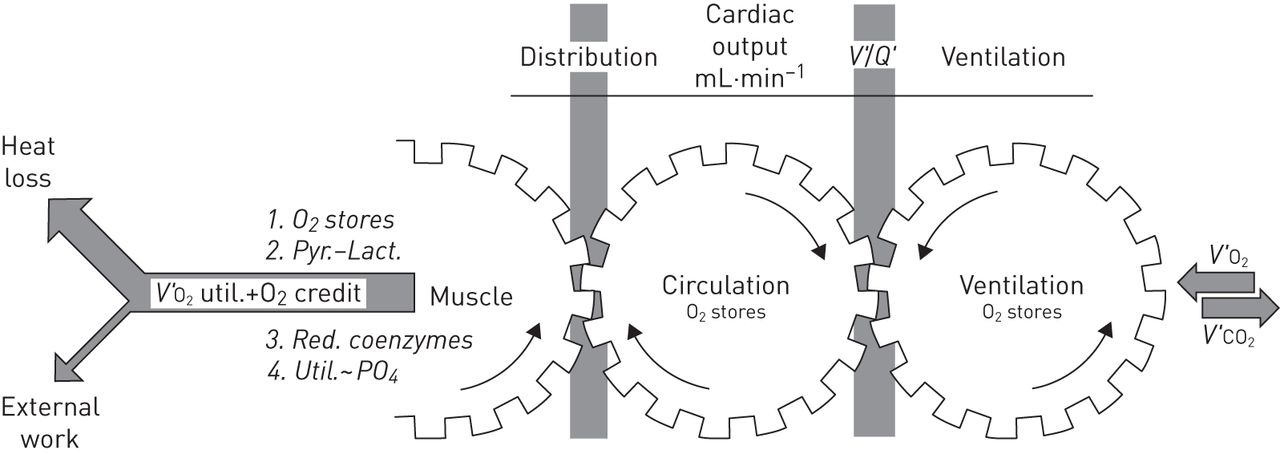
In order to appropriately interpret constraints that the ageing lung may pose for physical function, systemic adaptations associated with older age also demand consideration. Systemic integration of physiologic mechanisms underlying exercise response was described by Wasserman et al. [24] in 1967 (figure 2). Physiological systems directly involved in the response to maximal aerobic exercise include: 1) coupling of muscle metabolism to mechanical power output; 2) gas transport between muscle capillary and mitochondria; 3) matching of muscle blood flow to its requirement; 4) O2 and CO2 carrying capacity of the blood; 5) cardiac output; 6) pulmonary vascular function; 7) pulmonary O2 and CO2 transport; 8) control of ventilation; and 9) pulmonary mechanics and respiratory muscle function. This review will describe ageing-associated changes in each of these links, and how declining lung function may play a role in promoting multimorbidity in the elderly through limitation of physical function.
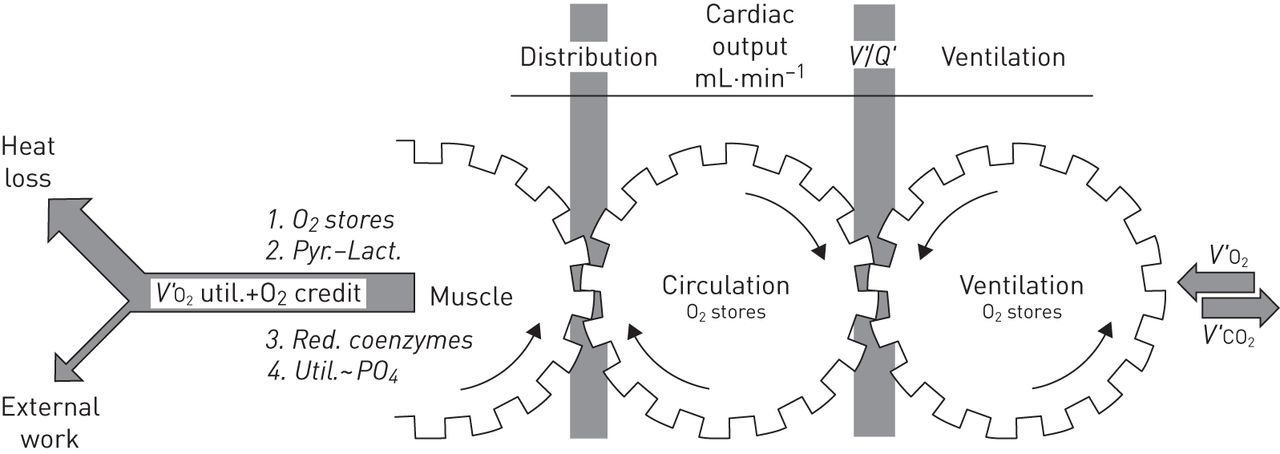
The interaction of physiological mechanisms during exercise based on the classic 1967 conceptualisation of Wasserman et al. [24]. The ability to perform exercise is dependent on the performance of a number of linked systems, each of which is subject to deterioration with ageing. V′O2: oxygen uptake; V′/Q′: ventilation/perfusion ratio; V′CO2: carbon dioxide production. Pyr.: pyrate; lact.: lactate; util.: utilisation. Reproduced from [24] with permission from the publisher.
Before embarking on an exploration of the effects of ageing on each of these physiological systems, it is worth stressing that cardiopulmonary exercise testing (CPET) can be a helpful clinical tool to evaluate the common complaint of dyspnoea on exertion in the elderly. CPET can often separate dyspnoea related to ageing from pathological causes. For example, potential contributors to increased dyspnoea in the elderly include incipient pathology, obesity, medications such as β-blockers and deconditioning. Reduced V′O2max and lactate threshold is seen in all these conditions, but distinguishing features may well be present. Obese subjects have increased V′O2 during unloaded exercise but preserved ΔV′O2/Δ power output. A blunted heart rate response to exercise can be seen in β-blockade. A reduced ΔV′O2/Δ power output slope on the other hand should trigger investigations for cardiovascular abnormality. Deconditioning and ageing often yield cardiopulmonary exercise test findings that are difficult to distinguish. These observations stress the importance of knowledge of the appropriate inter-relationships among the pulmonary–cardio–metabolic systems during exercise, and how these relationships may change with deconditioning and with age.
Coupling of muscle metabolism to mechanical power output
The transfer of metabolic to mechanical power output at rates necessary to meet sustained exercise task requirements can be usefully considered in three stages: 1) coupling of mitochondrial oxygen consumption to ATP production (mitochondrial coupling); 2) coupling of ATP hydrolysis to mechanical power production (mechanical coupling); and 3) economy of application of mechanical power to fulfil task requirements (biomechanic coupling or “skill”). Ageing influences each of these steps.
Muscle size, architecture and metabolism are altered with advancing adult age [25]. Limb muscles, particularly large locomotor muscles, are 25–35% smaller in older age and have more fat and connective tissue than those of younger individuals [26]. This decline is accompanied by a 30–40% decrease in muscle fibre number between the second and eighth decades [27]. Type II (fast-twitch) fibres are 10–40% smaller in the elderly, while type I (slow-twitch) fibre size is less affected. Remodelling of motor units associated with type IIx fibre loss [28], selective type IIa fibre denervation and collateral re-innervation of type I muscle fibres, results in type I fibre grouping in elderly muscle. This contributes to altered biomechanics of locomotor activity, reducing skill and increasing energy cost [29–32]. An additional component of skill deficit and muscle weakness in the elderly is disruption of excitation–contraction coupling, likely due to neuromuscular junction function loss, dihydropyridine receptor loss, impaired calcium release, and oxidative modification of myosin [33, 34]. Overall, these changes reduce available muscle mass for maximal aerobic exercise, and can lower economy of its application for external locomotion (impaired biomechanical coupling) [35].
Whether changes occur in mitochondrial and mechanical coupling of elderly muscles is more controversial. At the fibre level, total mitochondrial content tends to be reduced in both type I and II elderly fibres [36, 37]. Sensitisation to permeability transition and release of mitochondrial-derived pro-apoptotic factors may be responsible [38]. Importantly, reduced mitochondrial oxidative capacity is not fully reversible by endurance training after late middle age [39–41], and overall mitochondrial content in quadriceps muscle of 70–80 year olds, estimated using magnetic resonance spectroscopy, is correlated with V′O2max reduction [42]. Mild mitochondrial uncoupling of oxidative muscle fibres, possibly in response to age-associated oxidative stress increase, is proposed as a protective mechanism contributing to the relative longevity of the most active fibres [43]. It is unclear, however, whether reduction in mitochondrial coupling, observed in resting muscle, is maintained during exercise. Additionally, an ∼37% increase in ATP cost of power production (reduced mechanical coupling) is proposed in 70–79-year-olds based on magnetic resonance spectroscopic measurements of plantar flexion exercise [44], perhaps consequent to slower contractile relaxation, greater ATP cost of ion transport, and/or greater instantaneous stiffness (reduction in elasticity) in single fibres and whole muscles of elderly participants [45–47]. The effect of this mitochondrial and mechanical uncoupling, observed in small muscle groups, would increase the O2 cost of power production during exercise. This, however, appears not to be the case in cycle ergometry in elderly humans up to 80 years old, for whom O2 cost is unchanged compared with the young (∼10 mL min−1·W−1) [48]. Conversely, some males aged 70–79 years were found to have reduced ATP cost of force production during electrically evoked plantar flexion exercise [49] and eight, non-smoking, female centenarians, actually showed lower O2 cost of power production during incremental exercise than young controls [50]. These findings are consistent with improved mechanical or mitochondrial efficiency during exercise in the very old: an adaptation that may offset the influence of decreasing lung function with age [50]. However, ΔV′O2/Δpower output slope in incremental exercise is sensitive to both kinetics and efficiency of oxidative phosphorylation [51], and it seems plausible that slowed V′O2 kinetics in the elderly [52] may increase the contribution of substrate level phosphorylation to ATP provision (phosphocreatine breakdown and glycogenolysis accumulating lactate) during incremental exercise in these centenarians. Precise determination of mitochondrial and mechanical efficiency in the very old awaits resolution.
Overall, therefore, reduced mass and mitochondrial content in elderly locomotor muscles limit maximal power output and aerobic capacity and thus greatly reduce total ventilatory demand at V′O2max compared with younger individuals. Muscle fibrosis, reduced elasticity and reduced biomechanical coupling may, however, contribute to increasing locomotor activity ATP cost, and thus contribute to increasing ventilatory requirement at any given submaximal power output [53].
Gas transport between muscle capillary and mitochondria
Diffusive O2 transport between muscle capillary and mitochondria is an important site of limitation to maximal O2 flux [54–56]. Diffusive O2 conductance is dependent on several variables including muscle capillarity (specifically, area of apposition between capillary and fibre, and mean distance between capillary and mitochondrion over which O2 has to diffuse), capillary haematocrit (red blood cell volume contacting muscle capillaries), gas solubility (as influenced by muscle structural variations, such as increased lipid concentration that facilitates O2 diffusion), temperature and muscle myoglobin and mitochondrial concentrations. Of these, the main site of resistance to diffusive O2 flux in muscle is likely to be the distance between haemoglobin and muscle sarcolemma [57, 58]. Myoglobin spectroscopy measurements in exercising human muscle [59, 60] show a large drop in oxygen tension (PO2) between capillary blood and myocyte interior, highlighting both the large resistance to O2 flux at the capillary/fibre interface and the importance of maintaining high capillary PO2 to facilitate O2 diffusion.
In this context, recent re-analysis of data from the seminal 1966 Dallas Bedrest Study is pertinent [61]. Muscular and cardiovascular responses to exercise were established in five healthy 20 year-old men subjected to 3 weeks of supine bedrest followed by 8 weeks of high-intensity endurance exercise training [18], and reassessed after 40 years follow-up [17, 19, 20]. Longitudinal V′O2max decline (∼25%), accelerating after age 50, was similar to the decline seen following 3 weeks of bedrest in youth, and was associated with reductions in both convective O2 delivery (∼10% reduction in peak cardiac output, stroke volume, heart rate) and O2 extraction (∼18% reduction in arterio-venous O2 concentration difference). This age-associated reduction in V′O2max would also be sensitive to reductions in muscle O2 diffusing capacity [61], should muscle capillarity regression exceed decline in muscle oxidative capacity in older age. However, changes in human muscle capillarity in the elderly are equivocal: ∼10–30% lower capillary/fibre ratio in older muscles is common, either in cross-sectional studies or across 12 years of ageing between mid-60s to mid-70s [30, 62–64]. On the other hand, capillary geometry and heamatocrit, determined in young and old Fischer 344×Brown Norway hybrid rats, appear unaffected by ageing, with an increase in red blood cell flux compensating for the potential reduction in convective O2 delivery imposed by reduced capillarity, at least at rest [65]. During exercise, a lower microvascular PO2 in the elderly (either by fluorescence quenching in rat or near-infrared spectroscopy in human muscles) suggest that blood-to-tissue O2 movement may be impaired placing greater reliance on substrate-level phosphorylation during exercise [66–69]. However, the fractional contribution of glycogenolysis and phosphocreatine breakdown to total ATP production is similar during maximal aerobic exercise in muscle from old and young participants, though this was observed in plantar flexion exercise where relative perfusion (Q′) of active muscle is much greater than during locomotor exercise [44]. Therefore, considering that age-associated capillary rarefaction is somewhat less than the decrease in oxidative capacity of elderly muscle, it seems unlikely that anatomic size of capillary/fibre interface and O2 diffusional conductance plays a major role in limiting muscle aerobic performance in the elderly [70].
CO2 is approximately 20-times more diffusible than O2 in biological tissues, and high-capillary CO2 concentration may facilitate capillary oxyhaemoglobin unloading. Slowed kinetics of oxidative metabolism in the elderly require greater phosphocreatine breakdown for a given power output, and ensuing transient intramuscular alkalosis contributes to temporally slowing kinetics of CO2 output relative to its production [44, 49, 71, 72]. This, together with intra- and extra-muscular CO2 buffers, slows muscular CO2 production (V′CO2) kinetics compared with those of V′O2, and therefore may lessen ventilatory demands for CO2 clearance. Interestingly, expression of monocarboxylate lactate transporters 1 and 4, and the ratio of oxidative to glycolytic enzyme activity, are increased in older muscles, independent of physical activity [73, 74]. These adaptations may help ameliorate intramuscular acidosis and increase muscle fatigue resistance in high-intensity exercise in the elderly [75], but accelerate blood lactate appearance and onset of increased ventilatory demands associated with systemic metabolic acidosis [76]. Consequently, potential benefit from increased proton sequestration rate by phosphocreatine breakdown slowing CO2 flux towards the lung may be moderated by enhanced lactate and proton transport rate, contributing to driving ventilatory compensation for metabolic acidosis as V′O2max is approached. On balance, therefore, the influence of the aged muscle/capillary interface on exercise ventilatory demands is likely small.
Matching of muscle blood flow to its requirement
Although age-associated muscle microvascular anatomy changes may be less influential than reduced mitochondrial function in limiting maximal aerobic capacity in the elderly [70], there is strong evidence that muscle blood flow is attenuated during submaximal large muscle mass or locomotor exercise in older humans (∼55–80 years) [77, 78]. The locus of attenuated increase in active muscle blood flow in the elderly, which is accomplished through a combination of systemic sympatho-excitation and local metabolically mediated vasodilatation, is controversial. However, a sex difference may exist such that older oestrogen-deficient women are particularly prone to blunting of leg vasodilator responsiveness and more rapid muscle deoxygenation compared with younger controls [79–86]. Importantly, this disruption impairs the ability of elderly muscle to deliver O2 in appropriate proportion to its requirement (Q′/V′O2 ratio), which may contribute to transient microvascular deoxygenation during submaximal exercise and increased demand for substrate level phosphorylation [85, 87–89]. Reduced amplitude and kinetics of cholinergic, shear stress and endothelial-mediated relaxation, particularly in feed arteries of oxidative muscles, has been implicated [90, 91]; although the effect of older age on reactivity of muscle microvessels varies with branch order and vasoactive stimulus [92]. Response to metabolic dilators is also attenuated in the elderly, e.g. ATP-induced vasodilatation is lower in the sedentary elderly [93], although whether alterations in nitric oxide (NO) contribute to limiting exercise hyperaemia in the elderly is debated [77]. The metabolically-activated group III/IV muscle afferent contribution to exercise hyperaemia appears to be absent in older individuals, which may further impair blood flow distribution and reduce regional Q′/V′O2 in the elderly [94]. α1-Adrenergic vasoconstrictor tone appears to be similar between old and young, but may be less attenuated during exercise in the elderly simply due to lower absolute power outputs achieved [80].
While impaired exercise vasodilatation is observed in many elderly, lifelong physical activity protects against this effect [95]: only in sedentary elderly was limb muscle lactate release associated with an attenuated exercise hyperaemia. Interestingly, in men aged 62–73 years, where absolute locomotor muscle exercise hyperaemia was well preserved, a greater proportion of submaximal cardiac output was directed towards the legs at a given V′O2 compared with 20–25 year olds [84, 96]. This implies that competition for blood flow by other regional circulations could, at least as maximal cardiac output is approached, attenuate locomotor muscle blood flow rise in exercising older adults [97]. This may be particularly important in relation to competition for blood flow from respiratory muscles where greater deadspace ventilation and impaired pulmonary mechanics increases work of breathing for a given V′O2, and may contribute to limiting respiratory and/or locomotor muscle Q′/V′O2 in the elderly, as it can in athletes and patients with heart failure [98, 99].
Oxygen and carbon dioxide carrying capacity of the blood
Anaemia is prevalent in the ageing population with over 10% of individuals above age of 65 years affected [100]. Most cases of anaemia in older subjects are mild, but even mild decline in haemoglobin will decrease O2 carrying capacity and reduce V′O2max. Less is known about haemoglobin O2 affinity changes with ageing. A study of healthy male and female 18–89 year olds revealed an increase in the PO2 at which haemoglobin is half saturated (P50) in 60–89 year olds compared with 18–39 year olds, in keeping with age-related haemoglobin O2 affinity decrease. There was no significant change in 2,3-diphosphoglycerate concentration seen in this population [101]. Other work has shown that haemoglobin O2 affinity does not appear related to age in men [102]. There seems to be no data available regarding blood CO2 carrying capacity or buffering capacity changes with ageing.
Cardiac output
Maximal cardiac output decreases with age [12, 13]. Decline in maximal heart rate (∼0.7 beats·min−1·year−1 [103]) appears to be less severe than rate of V′O2max decline, suggesting that decreased sinoatrial node sensitivity to β-adrenergic stimulation in older individuals [104] is not a primary cause of aerobic capacity loss. Measurements of intrinsic heart rate, using intravenous infusions of propranolol and atropine to achieve autonomic blockade, reveal linear intrinsic heart rate decrease over ages 16–70 years [105]. Animal models show that the ageing sinoatrial node has decreased conduction velocity and contains fewer pacemaker myocytes [106]. Remaining sinoatrial myocytes demonstrate altered ion channel activity, leading to depressed excitability and consequently lower heart rate [106]. Similar changes may occur in ageing human sinoatrial myocytes, but further study is needed.
Ageing hearts may utilise a different mechanism to increase stroke volume during exercise than younger hearts. In elderly subjects, end-diastolic volume increases with exercise with minimal change in end-systolic volume; while in younger individuals, the increase in stroke volume with exercise is primarily due to a decrease in end-systolic volume [107]. Increasing end-diastolic volume may lead to larger stroke volume increase during exercise in older subjects, mitigating the influence of peak heart rate decline [107]. Age does not alter the cardiac output–V′O2 relationship although, for a given cardiac output, older subjects have lower leg blood flow [108].
Peak cardiac output falls ∼25% with age [12]. Despite significant structural heart changes with age [109–111], global left ventricle systolic function appears unaffected by healthy ageing [112–114]. Peak stroke volume also appears largely unchanged throughout life [111]. If one accepts a 50% drop in V′O2max from ages 20–80 years [12], 25% may be attributable to cardiac output decline. As stroke volume response to exercise is unchanged, reduced heart rate, as well as maldistributed cardiac output (as discussed earlier), are responsible for the cardiac contribution to age-related decline in V′O2max. The exercise cardiac response of the ageing individual has been likened to that of a young person on β-blockers [107].
Pulmonary vascular function
Ageing-related pulmonary circulation changes influence exercise response in the elderly subject. Pulmonary vascular stiffness increases with age [115, 116]. Decreased pulmonary vascular compliance, along with decreased left ventricle compliance [114], leads to increased pulmonary arterial pressure, pulmonary wedge pressure and pulmonary vascular resistance in older individuals [117, 118]. Pulmonary arterial pressure increase appears to be secondary to vascular stiffening and decreased left ventricle compliance [118].
In a recent right heart catheterisation study, subjects older than 55 years showed resting haemodynamics similar to those of younger individuals. However, significant differences developed during exercise. The older group displayed lower cardiac output and greater mean pulmonary arterial pressure. Increased mean pulmonary arterial pressure during exercise with advancing age was the consequence of increased pulmonary vascular resistance and elevated left ventricle filling, due to age-related diastolic dysfunction [119].
Pulmonary oxygen transport
The assumption that arterial O2 partial pressure (PaO2) declines at a constant rate between ages 20 and 100 years is founded on prediction equations based on a small number of individuals above age 60 years [120]. These equations may underestimate values for elderly subjects, with a wide prediction range of 63–84 mmHg for an 82-year-old subject [120]. There is evidence to the contrary: Blom et al. [121] reported a plateau in PaO2 decline after age 70 years. Similarly, other recent studies show age-related decline in PaO2 between ages 40 and 74 years with no significant association between PaO2 and age greater than 70 or 74 years [122–124]. There is potential for survival bias in these results, however, as individuals with lower PaO2 may die earlier [125, 126]. There are also sex differences for PaO2 in the elderly population. A well-done study by Hardie et al. [123] demonstrated a mean PaO2 of 77 mmHg (lower 95% confidence limit of 62 mmHg) and 73.5 mmHg (lower 95% confidence limit of 59.6 mmHg), respectively, for men and women over age 70 years.
PaO2 changes with ageing may be mechanistically related to gas diffusion properties of the lung and also to ventilation-perfusion distribution. There appears to be little effect on exercise gas exchange capabilities in older individuals, as exercise-induced arterial hypoxaemia is infrequent. However, exercise-induced hypoxaemia occurs more frequently in highly fit elderly individuals [23]. The mechanism is not known, but plausibly may be related to physiological changes noted above, and the high power outputs achieved in fit individuals (and therefore greater cardiac output and reduced capillary transit time), compared with elderly subjects of average fitness. As discussed below, increased alveolar deadspace and increased alveolar ventilation (V′) to pulmonary perfusion (V′/Q′) mismatch in older subjects likely contribute to exercise-induced hypoxaemia seen in elderly athletes.
Pulmonary trans-capillary gas diffusion
Ageing leads to decreased capacity for pulmonary gas exchange, reflected in decline in diffusing capacity of the lung for carbon monoxide (DLCO) [127]. DLCO decline may be in part related to gradual reduction in alveolar–capillary density to alveolar diameter ratio in the older lung, along with decreased pulmonary capillary blood volume and increased V′/Q′ mismatch that are seen in the elderly [128].
Matching of ventilation to perfusion
Smaller studies have demonstrated that ageing results in an increase in lung areas with high V′/Q′ (physiological deadspace) and low V′/Q′ (shunt) [129, 130]. Older subjects have shown increased alveolar to arterial PO2 difference (PA–aO2) [131, 132]. PA–aO2 can be widened by development of right-to-left shunting, diffusion limitation, or V′/Q′ mismatch. Cardus et al. [133] attempted to determine whether increasing V′/Q′ mismatch with age causes age-related PaO2 decline and found a small PaO2 decrease with age (6 mmHg between ages 20 and 71 years) that was explained by a small V′/Q′ mismatch increase. Increased intra-pulmonary shunting (low V′/Q′) did not appear to contribute to lower PaO2. Unfortunately, this was a relatively young population: only four of 64 subjects were above age 60 years. As closing volume does not equal functional residual capacity until the age of 65 years [134], it is possible there may be additional low V′/Q′ (shunt) units in more elderly subjects.
Distribution of ventilation
Deadspace to tidal volume ratio (VD/VT) is elevated at rest in older individuals. VD/VT decreases with exercise but the nadir value is higher in older than in younger subjects. In young athletes, maximal exercise VD/VT averages 13%, while in older subjects, VD/VT averages 30% [135].
Ventilation is primarily distributed to the lower lung in younger subjects [136]. Xenon distribution measurement of elderly lungs reveals that in older lungs all airways are open above 65% of total lung capacity [137]. Electrical impedance tomography of aged lung demonstrates absence of posture-dependent changes in gas distribution normally seen in younger lungs [138]. As a consequence, at resting tidal volumes, ventilation to dependent lung is decreased in older individuals, leading to greater ventilation of the upper lung and increased upper lung perfusion that improves V′/Q′ matching.
Control of ventilation
The respiratory control system adjusts minute ventilation (V′E) to respond to changes in metabolic rate and other perturbations in order to maintain, as much as possible, arterial homeostastis. Resting pulmonary ventilation is adjusted to regulate PaCO2 (and thus arterial pH) within a narrow range. PaO2 only becomes an appreciable ventilatory stimulus when PaO2 drops well below the normal range. Though few systematic studies have been reported, at rest the elderly appear to regulate PaCO2 within the same range as the young; PaO2 is somewhat lower mostly because of increased V′/Q′ inhomogeneity [53, 139].
Challenges to the respiratory control system include exercise, inhalation of hypercapnic and hypoxic mixtures and resistive and elastic loads to breathing. Of these, exercise is the most commonly encountered challenge and has, therefore, received the most study. Interestingly, though, in comparison with the relatively preserved functional characteristics of exercise ventilatory control (discussed later), the elderly exhibit substantial degradation of response to these other challenges. Response to inhaled CO2 is blunted [139–143]; Brischetto et al. [140] found that the V′E–PaCO2 slope was almost one-third lower in the elderly. Similarly, hypoxic response is reduced in older individuals [141–143]; Peterson et al. [143] found hypoxic ventilatory response to be reduced about 50%. Responses to both resistive and elastic loaded breathing are also reduced [144, 145].
Alterations in exercise ventilatory response are more subtle. A consistent observation is that ventilatory response to exercise at a given V′CO2 is elevated in elderly subjects as compared to the young [76, 140, 146–148]. Inbar et al. [147] reported cardiopulmonary responses to incremental exercise of 1424 men, 43 of whom were aged 60–70 years. V′E/V′CO2 was distinctly greater across metabolic rates in the older group. Similarly Poulin et al. [148] studied the incremental treadmill exercise response in 128 men and 96 women aged 55–86 years. On average, the V′E/V′CO2slope was 12.3% greater per decade in men and 9.3% greater per decade in women.
The source of enhanced ventilatory response can be evaluated by considering the alveolar mass balance equation: V′E/V′CO2=k/(PaCO2×(1−VD/VT)), where k is a constant. This equation dictates that the greater V′E/V′CO2 can have only two sources: lower PaCO2 or greater VD/VT. Brischetto et al. [140] sampled arterial blood serially during incremental exercise in two older subjects and found an isocapnic response. Mummery et al. [149] drew arterial blood samples at rest and after 6 min of moderate and heavy exercise from 10 older (average age 63 years) and 10 young subjects. Moderate exercise was isocapnic, and PaCO2 fell with heavy exercise (presumably in response to metabolic acidosis), with no differences between older and younger participants. Calculations using measurements of PaCO2 suggest that deadspace is greater in elderly subjects than in young [53, 149]. A review of the literature for the source of elevated deadspace ventilation reveals only inconsistent alterations in the tidal volume-breathing frequency relationship [76]. Therefore, deadspace ventilation elevation during exercise seen in the elderly seems likely related to increased alveolar deadspace.
In contrast to moderate intensity steady-state exercise responses, dynamic exercise ventilatory responses are distinctly modified in the elderly. In studies of young subjects, dynamic response of V′E is closely correlated with V′CO2 dynamics [150, 151], with the result that PaCO2 fluctuation during the dynamic phase of exercise is small [151]. The observation that V′E kinetics are substantially slowed in the elderly (with response time constants averaging 40–56% greater than in young subjects) [152, 153] might suggest that PaCO2 regulation in the non-steady state is greatly degraded, but this is likely not the case. The key observation is that V′O2 kinetics are slowed in the elderly [152–154]. This is primarily related to low muscle oxidative capacity in ageing, but may also be influenced by wider muscle Q′/V′O2distribution (see above). V′O2 kinetics are the prime determinant of V′CO2 kinetics, with the latter being slowed with respect to the former by muscle-alkalinizing effects of phosphocreatine breakdown and fluctuation in the body's large CO2 stores [155]. Thus, V′CO2 kinetics are also markedly slowed in the elderly [152, 153]. Importantly, the ratio of V′E and V′CO2 time constants are somewhat greater in elderly compared to younger subjects [152, 153], implying a slightly “looser” control of PaCO2. An important observation from eight older subjects (aged 65–78 years) undergoing a rigorous exercise-training programme was that training speeded V′O2, V′CO2 and V′E kinetics (each time constant was reduced by ∼50%), with correlation between the change in V′E and V′CO2 time constants being strong (r=0.65) [152]. This supports the concept that slower V′E kinetics in the elderly are mostly related to slowed metabolic rate kinetics, but a small degradation of PaCO2 control cannot be excluded.
Pulmonary mechanics and respiratory muscle function
With age, lung structural changes occur that affect exercise ventilatory response. These changes have the potential to contribute to decreased exercise performance and decline in V′O2max seen with increasing age [10, 12, 147, 156, 157]. The lung's ageing process is difficult to generalise, given differences in individual environmental and genetic factors that influence how an individual ages. Sex differences in lung ageing may also be significant. The increase in resting lung and residual volumes and reduced vital and inspiratory capacities that occur with age have been well described in the literature and are beyond this review's scope [139, 158–161].
Pulmonary function begins to decline at approximately age 25 years. In healthy non-smoking individuals, spirometric measures forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) decrease by ∼30 mL·year−1 in men and 23 mL·year−1 in women, with accelerated loss after age 65 year [162, 163], Mean bronchial diameter also decreases with age, yielding increased airway resistance, particularly in peripheral airways [164]. Pulmonary static elastic recoil pressure decreases by approximately 0.1–0.2 cm·year−1 after age 20 years due to chest wall stiffness increase and lung tissue elasticity loss [165, 166]. Elasticity loss is thought to represent remodelling of both spatial arrangement and cross-linking of the lung's elastin-collagen network [167]. Elastic recoil loss leads to alteration in the expiratory portion of the maximal flow-volume loop and results in the characteristic “scalloped” loop seen in elderly non-smokers. Elastic recoil loss appears proportional to peak expiratory flow decrease with age [168].
Over time, chest wall compliance decreases due to calcification of costal cartilage, with increased prevalence of both spinal kyphosis and osteoporosis-associated vertebral fractures potentially contributing [169–171]. Obesity is prevalent in the ageing population, affecting more than one-third of adults older than 65 years [172]. Obese individuals have lower respiratory system compliance [173–175]. Expiratory reserve volume clearly decreases with increasing body mass index; functional residual capacity is reduced to a lesser extent. Total lung capacity does not appear to be affected significantly, except in extreme obesity. Decreased compliance is expected to contribute to increased dyspnoea during exercise in obese individuals [176, 177].
Respiratory muscle strength decreases with age [178, 179]. Maximal effort transdiaphragmatic pressure gradients in older individuals are lower than in younger subjects, reflecting decreased diaphragmatic strength [180, 181]. In one study, diaphragm strength in the elderly was 13% less than in a younger group by maximal sniff and 23% less using cervical magnetic stimulation. Other measurements have shown 25% lower diaphragmatic strength in the elderly [181]. Strength loss does not appear related to diaphragmatic fibre type change or muscle atrophy [182–184]. Diaphragm and intercostal muscle stiffness increase with age [185], decreasing chest wall compliance. Diaphragm collagen metabolism changes (collagen concentration and cross-linking increases) appear responsible for increased stiffness [186]. Spinal kyphosis and increased chest anterior–posterior diameter that occur with age likely contribute to decreased diaphragmatic function [158].
Airspace size increases with age [187]. The term “senile emphysema” that has been used to describe this age-related pulmonary morphological change is inaccurate; although ageing results in alveolar duct enlargement and distal duct ectasia, the ageing lung does not develop alveolar wall destruction and inflammation that is a hallmark of smoking-related emphysema [188].
In healthy adults, peak ventilation during exercise typically approaches 70% of measured maximal voluntary ventilation, demonstrating appreciable breathing reserve. Ageing is associated with a greater V′E and dyspnoea for a given power output (figure 3) due to mechanisms discussed earlier, including reduced lactate threshold, and increased VD/VT and V′/Q′ mismatching (likely reflected in the increased V′E/V′CO2). Breathing reserve tends to decrease in athletes and with normal ageing [189]. In a recent study of 759 maximal treadmill exercise tests performed in healthy Norwegian adults aged 20–85 years, peak ventilation decrease started at ages 40–49 years [190]. While peak V′E decreased, predicted maximal voluntary ventilation (based on both FEV1·35 and FEV1·40) [156, 191] also decreased due to age-related FEV1 reduction. This led to breathing reserve preservation. There is also an age-related decline in vital capacity that leads to relative limitation in tidal volume (figure 3), meaning, for a given ventilation, breathing frequency tends to be greater, especially at higher intensities [161, 192].

{kind=link}
{kind=link}
{kind=link}
Perceptual, ventilatory and respiratory mechanical responses to incremental treadmill exercise in healthy older (OM; aged 60–80 years) compared with younger men (YM; aged 40–59 years). Data are presented as mean±sem for measurements at rest, during each stage of exercise and at peak exercise. V′O2: oxygen uptake; V′E: minute ventilation; V′CO2: carbon dioxide production; VT: tidal volume; IRV: inspiratory reserve volume; TLC: total lung capacity; IC: inspiratory capacity. Reproduced from [161] with permission from the publisher.
During exercise in youth, tidal volume increase is achieved through decreases in both inspiratory and expiratory reserve volumes [193, 194]. There is initially a drop in end-expiratory lung volume (EELV) in both young and old subjects to optimise inspiratory muscle function [195]. During severe intensity exercise in fit, young athletes, expiratory flow limitation can develop [193], although increased EELV does not appear until very high rates of ventilation [23, 195]. Airway diameter decrease and static recoil pressure reduction that occur in ageing suggest that flow limitation may also become a factor limiting exercise in older individuals. Accurate measurement of expiratory flow limitation during exercise is challenging. The most common method involves demonstrating impingement of exercise flow–volume loops on the maximum-effort resting expiratory flow–volume relationship. This method, however, does not account for thoracic gas compression and may overestimate flow limitation during exercise [192, 196]. Taking several expirations at variable efforts from total lung capacity to residual volume may help correct for dynamic gas compression [196]. Using a post-exercise maximal expiratory flow–volume curve also helps by accounting for exercise-induced bronchodilation [192]. Wilkie et al. [195] reported that older women exhibit exercise expiratory flow limitation more frequently than younger women. Older subjects also report greater breathlessness and a steeper slope of the dyspnoea–power output relationship, suggesting that age-related lung changes have symptomatic consequences (figure 3). Interestingly, this study found an EELV increase at power outputs between 80–100% V′O2max only in younger women. The authors hypothesise that impending flow limitation in older women may have an effect on EELV regulation [195]. This contrasts with findings of another study of trained individuals (14 men, four women aged 62–82 years) where older subjects increased EELV during moderate intensity exercise, but EELV did not increase in younger subjects until V′E exceeded 110–120 L·min−1 [23]. Increased EELV compromises operating length–tension relationships of diaphragm and respiratory muscles, leading to less force generation capacity. Increased EELV during exercise results in the subject breathing on the flattened portion of the lung pressure–volume relationship, reducing inspiratory muscle length, increasing work of breathing and potentially decreasing inspiratory muscle endurance [192]. In line with these effects, inspiratory reserve volume is consistently lower in the elderly at rest, and remains lower, along with inspiratory capacity, for any given level of V′E compared with young subjects (figure 3), likely contributing importantly to the greater sensation of breathlessness in the elderly [161, 197, 198].
While flow limitation and EELV behave in a similar fashion during low intensity exercise in older and younger lungs, expiratory flow limitation seems to develop at lower intensity exercise in older subjects [23, 53, 195]. There may be a sex difference, with women developing expiratory flow limitation more frequently than men during high intensity exercise [199], presumably related to decreased lung size and lower maximal expiratory flow rates in women. Guenette et al. [200] describe an 86-year-old female lifelong competitive swimmer (former Olympian) with moderate airflow obstruction (FEV1/FVC 53%; FEV1 54% predicted) who continued regular exercise into old age. Despite severe ventilatory limitation (dynamic hyperinflation of 780 mL and end inspiratory lung volume of 96% total lung capacity) the participant only reported moderate dyspnoea and achieved V′O2max of 175% predicted (19.6 mL kg−1 min−1). The authors speculate reduced ventilatory requirements, breathing pattern alterations and improved respiratory muscle strength may each contribute to reduced dyspnoea in this athletic octogenarian. While only a case report, these findings emphasise that relative preservation throughout life of aerobic capacity may be possible with regular high-intensity exercise, even when expiratory flow and ventilatory limitation is present.
Conclusions
Maintaining a high level of physical activity is an important part of healthy ageing and minimisation of multimorbidity. Deterioration in various components of the multi-organ system response to exercise in the elderly conspires to make this difficult. Decreases in pulmonary system function likely contribute to exercise intolerance in healthy elderly, particularly those who maintain physical activity into senescence. However, loss of muscle oxidative capacity and cardiac output in sedentary elderly outstrips decline in pulmonary function, such that the relatively small contribution of pulmonary function to exercise limitation is preserved over a wide range of ages. Training programmes for muscles of ambulation remains the most effective way to retain aerobic capacity in older individuals. However, the degree to which maintenance of training past 70 years of age, which is associated with considerable health-benefits, causes encroachment upon pulmonary limits requires further study.
Acknowledgements
R. Casaburi holds the Grancell/Burns Chair in the Rehabilitative Sciences.
Footnotes
Previous articles in this series: No. 1: Faner R, Cruz T, López-Giraldo A, et al. Network medicine, multimorbidity and the lung in the elderly. Eur Respir J 2014; 44: 775–788. No. 2: Divo MJ, Martinez CH, Mannino DM. Ageing and the epidemiology of multimorbidity. Eur Respir J 2014; 44: 1055–1068. No. 3: MacNee W, Rabinovich RA, Choudhury G. Ageing and the border between health and disease. Eur Respir J 2014; 44: 1332–1352. No. 4: Carraro S, Scheltema N, Bont L, et al. Early-life origins of chronic respiratory diseases: understanding and promoting healthy ageing. Eur Respir J 2014; 44: 1682–1696. No. 5: Chacko A, Carpenter DO, Callaway L, et al. Early-life risk factors for chronic nonrespiratory diseases. Eur Respir J 2015; 45: 244–259. No. 6: Barnes PJ. Mechanisms of development of multimorbidity in the elderly. Eur Respir J 2015; 45: 790–806. No. 7: Rodriguez-Roisin R, Bartolome SD, Huchon G, et al. Inflammatory bowel diseases, chronic liver diseases and the lung. Eur Respir J 2016; 47: 638–650. No. 8: Spagnolo P, Cordier J-F, Cottin V. Connective tissue diseases, multimorbidity and the ageing lung. Eur Respir J 2016; 47: 1535–1558.
Conflict of interest: None declared.
- Received February 15, 2016.
- Accepted June 27, 2016.
- Copyright ©ERS 2016
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Coupling of muscle metabolism to mechanical power output
- Gas transport between muscle capillary and mitochondria
- Matching of muscle blood flow to its requirement
- Oxygen and carbon dioxide carrying capacity of the blood
- Cardiac output
- Pulmonary vascular function
- Pulmonary oxygen transport
- Control of ventilation
- Pulmonary mechanics and respiratory muscle function
- Conclusions
- Acknowledgements
- Footnotes
- References
- Figures & Data
- Info & Metrics