Article Text

Abstract
Background Primary ciliary dyskinesia (PCD), a genetically heterogeneous condition enriched in some consanguineous populations, results from recessive mutations affecting cilia biogenesis and motility. Currently, diagnosis requires multiple expert tests.
Methods The diagnostic utility of multigene panel next-generation sequencing (NGS) was evaluated in 161 unrelated families from multiple population ancestries.
Results Most (82%) families had affected individuals with biallelic or hemizygous (75%) or single (7%) pathogenic causal alleles in known PCD genes. Loss-of-function alleles dominate (73% frameshift, stop-gain, splice site), most (58%) being homozygous, even in non-consanguineous families. Although 57% (88) of the total 155 diagnostic disease variants were novel, recurrent mutations and mutated genes were detected. These differed markedly between white European (52% of families carry DNAH5 or DNAH11 mutations), Arab (42% of families carry CCDC39 or CCDC40 mutations) and South Asian (single LRRC6 or CCDC103 mutations carried in 36% of families) patients, revealing a striking genetic stratification according to population of origin in PCD. Genetics facilitated successful diagnosis of 81% of families with normal or inconclusive ultrastructure and 67% missing prior ultrastructure results.
Conclusions This study shows the added value of high-throughput targeted NGS in expediting PCD diagnosis. Therefore, there is potential significant patient benefit in wider and/or earlier implementation of genetic screening.
- primary ciliary dyskinesia
- mutation spectrum
- cilia
- bronchiectasis
- population
Statistics from Altmetric.com
Introduction
Primary ciliary dyskinesia (PCD) is a rare genetic disease caused by cilia dysmotility that is associated with a range of defects of motile cilia structure and biogenesis. PCD is typically an autosomal or X linked recessive disorder caused by mutations in >40 different genes encoding structural ciliary proteins, cilia assembly, and transport factors and proteins implicated in multiciliogenesis.1 2 Children and adults affected by PCD consequently manifest with progressive respiratory disease characterised by bronchiectasis and impaired lung function. Symptoms often present in early life with neonatal respiratory distress syndrome and persist with chronic wet cough, rhinitis, sinusitis, otitis media and hearing defects.3 Defective cilia of the brain ependyma, fallopian tubes and developing embryo can explain other disease features. Half of patients have laterality defects arising from embryonic nodal cilia dysfunction, and a significant proportion of male patients are subfertile with defective sperm flagella. Affected individuals, in particular those with reduced cilia numbers, can also manifest with hydrocephalus, while RPGR and OFD1 mutations can, respectively, cause rare retinal dystrophy and oral-facial-digital syndrome PCD subtypes.1 2 4 5
The prevalence of PCD is around 1:15 000 worldwide. PCD occurs much more frequently in highly consanguineous communities such as the UK South Asian population, in whom disease prevalence is as high as 1:2300.3 Generally, PCD symptoms are variable and diagnosis is frequently delayed or missed.6 Early diagnosis has potential to improve morbidity since lung damage can be delayed by specialist care.3 7 PCD diagnostic testing requires access to a combination of investigations including measurement for low nasal nitric oxide levels, high-speed video microscopy for ciliary beating defects, ciliary ultrastructure defects analysed by transmission electron microscopy (TEM), immunofluorescence staining for abnormal motile cilia proteins, and increasingly genetic analysis.7 8
PCD genetic diagnosis requires the identification of biallelic autosomal or hemizygous X linked mutations.7 8 Mutations in known PCD genes have been found in 60%–70% of tested patients with PCD.1 3 With additional genes still to be identified, the sensitivity of genetic testing as a ‘gold standard’ diagnostic test is reduced. However, with progressive identification of the whole ‘morbid genome’ causing PCD and ongoing reductions in DNA sequencing costs, genetics can increasingly be considered as a first-line test in the diagnostic pathway. Gene panels can currently be more effective for target sequence coverage and reduced time and costs than whole exome or genome sequencing.9
Here, we present a targeted next-generation sequencing (NGS) gene panel approach for characterisation of a multiancestry cohort of patients with PCD. Our aim was to investigate the utility of this approach for PCD, a clinically and genetically heterogeneous condition where current diagnosis requires multiple expert tests.8
Materials and methods
Subjects
One hundred and sixty-one unrelated families confirming self-reported ancestry and consanguinity at time of recruitment were ascertained from UK national PCD diagnostic and management services (London Royal Brompton Hospital, University Hospital Southampton, Leeds General Infirmary, Bradford Royal Infirmary, Birmingham Children’s Hospital and Leicester Royal Infirmary) and collaborating clinical centres in Portugal (Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisbon), Palestine (Makassed Hospital, East Jerusalem) and Egypt (Alexandria University Children’s Hospital, Alexandria). Recruitment took place between January 2015 and February 2017. The diagnosis of PCD followed the European Respiratory Society guidelines,8 using various methods according to the clinical centre, including clinical presentation and the results of formal PCD diagnostic tests (nasal nitric oxide level, cilia ultrastructure analysis by TEM, cilia beat pattern and frequency by high-speed video microscopy and immunostaining against specific ciliary proteins). Study inclusion criteria were based on a clinical suspicion of PCD and/or available cilia ultrastructural TEM analysis. TEM data were not available in a total of 27 families, who were included in the study based on other clinical criteria suggesting PCD.
Targeted NGS
Genomic DNA extracted from whole blood samples or saliva was screened for mutations using targeted NGS gene panels containing all the known PCD (online supplementary table S1) and isolated heterotaxy genes, plus one of two iterations of a larger set of cilia motility-associated candidate genes. These were collated after extensive literature searches for candidates with cilia involvement confirmed or likely, and from data from previous human genetics and PCD model organisms studies. Panel probe design used the Agilent SureDesign tool (Agilent Technologies, Santa Clara, California, USA) to capture all coding regions and 25 bp at the exon–intron boundaries (online supplementary tables S1 and S2). Capture probes were enriched in regions with potential low coverage. The SureSelectQXT NGS target enrichment kit (Agilent Technologies) was used for library preparation following the manufacturer’s protocol. Paired end sequencing (2×150 bp) was performed using the NextSeq 500/550 High Output V.2 Kit and NextSeq sequencing platform (Illumina, California, USA). Multiplexing of 48 samples was done on the same flow cell per sequencing run. Sequencing data were processed using an inhouse bioinformatics pipeline at North East Thames Regional Genetics Service.10 Variants were filtered for significance to produce variant lists of interest in each patient that conform to the expected minor allele frequency for PCD (<1%) and an autosomal or X linked recessive inheritance pattern. Variants were prioritised based on their minor allele frequency in the ExAc database,11 1542 individuals in the Born-in-Bradford cohort of UK South Asians12 and the al mena database of genetic variants in Middle East and North African individuals.13 Potential pathogenicity was assessed using several software including Human Splicing Finder, SIFT, PolyPhen-2, MutationTaster and Combined Annotation Dependent Depletion score. Variant pathogenicity scoring was done according to the guidelines of the American College of Medical Genetics and Genomics (online supplementary figure S1),14 using a well-established classification (or tiering) system of predicted pathogenic, likely pathogenic, uncertain significance variants versus likely benign or benign variants. Details of all variants of interest with the gene transcript numbers are in online supplementary table S3. For all affected individuals, a search for large insertion/deletion mutations and CNVs (using ExomeDepth software) was separately performed.15
Supplemental material
Sanger sequencing
All prioritised variants were confirmed in the proband and segregated within the available family members using Sanger sequencing. Sequencing data were viewed using SnapGene (GSL Biotech, Chicago, USA) or Sequencher software (Gene Codes Corporation, Ann Arbor, Michigan, USA).
Results
Targeted NGS yields high diagnostic output in a multiancestry cohort of patients with PCD
The probands from 161 unrelated families were screened using targeted NGS multigene panel analysis, followed by Sanger sequencing-based segregation analysis to confirm all identified genetic variants of interest and determine their familial inheritance pattern. All families had affected individuals with a suggestive clinical phenotype, in addition to either (1) a ciliary ultrastructural defect confirmed (97 families); or (2) inconclusive TEM results where PCD was still highly suggestive (37 families); or (3) no diagnostic TEM analysis yet performed but PCD still clinically highly suspected (27 families). Their details are summarised in table 1. The PCD-consistent features of the total 27 families with probands lacking TEM data are summarised in online supplementary table S4. The ancestry and consanguinity of all the families are summarised in online supplementary table S5, showing that 46% (74) were European, 22% (35) South Asian, 18% (29) Arab and the rest had other ancestries. Consanguinity was reported in 29% of 161, with highest levels in the Arab (25, 86%) and South Asian (12, 34%) families.
Genetic stratification of 161 unrelated PCD families, according to TEM findings
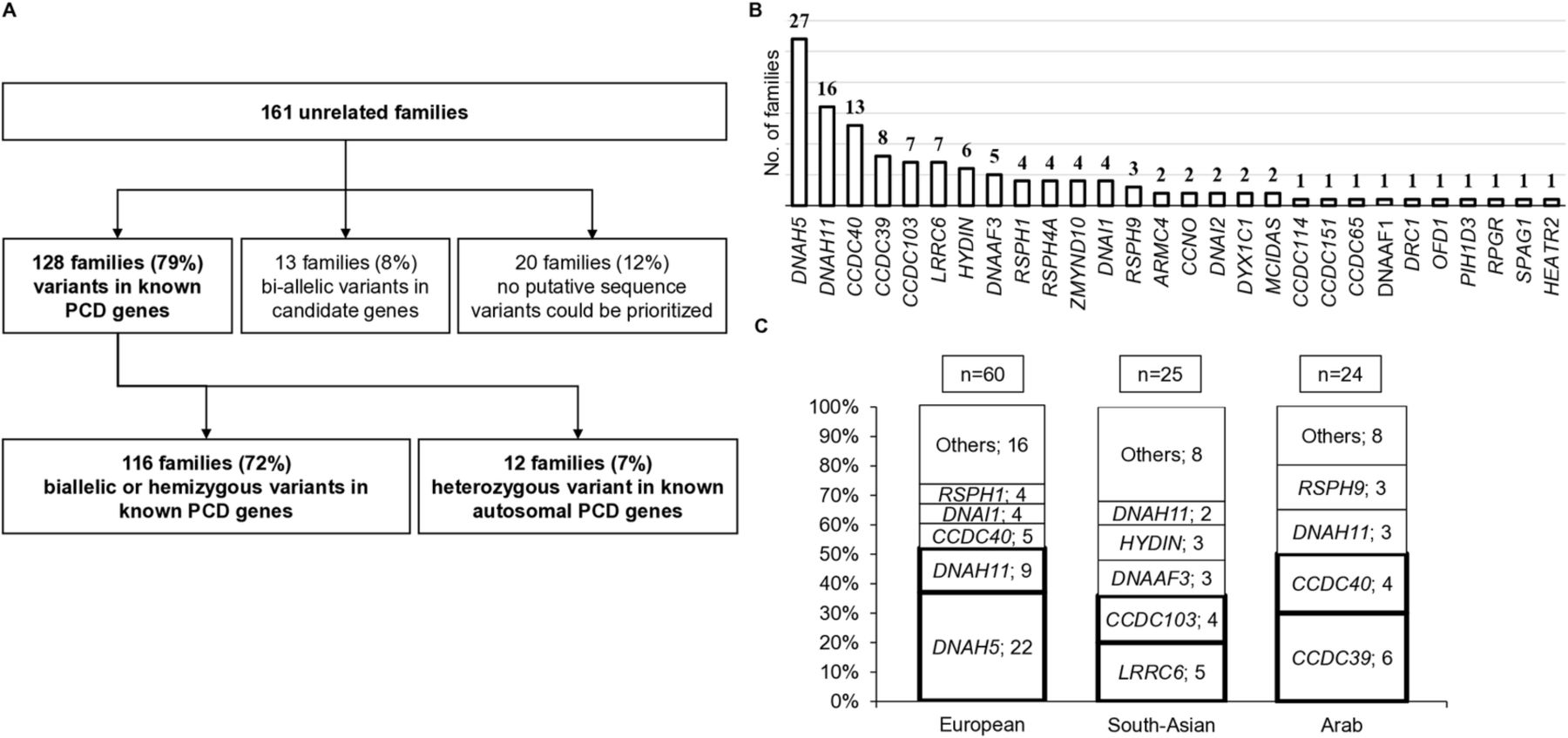
We identified causal variants in known PCD genes initially in 128 out of the 161 PCD families, then subsequently 4 more due to candidates being published as PCD genes, thus comprising finally 82% of the cohort (figure 1A and online supplementary table S3). Biallelic autosomal or hemizygous X linked variants were identified in 116 families in known PCD genes, with the further 4 families comprising 2 with CFAP300 variants16 and 2 with DNAH9 variants.17 Since these genes have been recently verified as PCD-causing, this means that a total of 120 out of 161 (75%) families were diagnosed. In 12 (7%) families, only one mutant allele (single heterozygous) was found in a known autosomal PCD gene, which is considered an incomplete genetic diagnosis. However, we include the variant data for all 12 families, since seven are protein truncation variants and five are already reported in previous studies on patients with PCD; also, all these ‘single hit’ variant-carrying patients had cilia ultrastructural defects consistent with the implicated mutant gene (figure 1A and online supplementary table S3).

Targeted next-generation sequencing yields a high (72%) diagnostic output in patients with PCD and reveals a diverse mutation landscape stratified by ancestry. (A) Flow chart of genetic results found for families enrolled in the study describing the genetic diagnostic output. Probands from 161 unrelated families were subject to next-generation sequencing using a multigene motile ciliome panel. Affected individuals in 4 of 13 ‘candidate gene’ families carried biallelic variants in two genes now recognised as disease-causing, CFAP300 and DNAH9; see the main text for details, this increases the diagnostic yield to 82%, rather than 79%. (B) The genetic stratification of all 161 families from the multiancestry cohort found to carry mutations in known PCD genes. (C) Summary of mutated genes in three different populations illustrating that different ancestries have different genetics. Among 109 European, Arabic and South Asian families, the genes most commonly detected to carry mutations are DNAH5 in European families (37%), LRRC6 and CCDC103 in South Asian families (36%), and CCDC39 and CCDC40 in Arabic families (42%). PCD, primary ciliary dyskinesia.
For 13 (8%) families, biallelic variants in candidate genes for PCD (CFAP300, DNAH9 included at the time) were identified, and further functional characterisation of these genes and their roles in causing PCD is ongoing. Finally, 20 (12%) families had no putative significant sequence variants, 11 of these having cilia ultrastructural defects identified by TEM and 3 having low nasal nitric oxide and abnormal cilia beat frequency but inconclusive TEM (online supplementary figure S2), and the other 6 having a strong clinical suspicion of PCD (situs inversus and recurrent respiratory problems) without prior investigations.
Significant, population-based genetic stratification underlies PCD
Prioritised variants for the 128 diagnosed families were identified within different functional categories of known PCD genes (figure 1B). The most prevalent, identified in 38% of families, affected genes encoding outer dynein arm (ODA) components (DNAH5, DNAH11, DNAI1, DNAI2). The second collectively most common affected genes encode dynein assembly factors (LRRC6, DNAAF3, ZMYND10, DYX1C1, DNAAF1, PIH1D3, SPAG1, HEATR2) in ≈17% of families, followed by mutations in ‘ruler protein’ genes (CCDC39, CCDC40) in 16% and radial spokes (RSPH1, RSPH3, RSPH4A, RSPH9) in 8% of families. CCDC103 mutations affected 5% of families, but otherwise mutations in genes involved in ODA docking (ARMC4, CCDC114, CCDC151), central pair (HYDIN) and nexin-dynein regulatory complex structures (CCDC65, DRC1), multiciliogenesis (CCNO, MCIDAS), or causing ‘syndromic’ forms (OFD1, RPGR) were more rare, affecting collectively ≈9% of the families.
Overall, DNAH5 was the most prevalent mutant gene, mutations identified in affected individuals from 27 (21%) families (figure 1B and detailed in figure 2A). However, populations of different ancestry (ethnicity) had considerably different genetic profiles. DNAH5 and DNAH11 mutations were found in 37% and 15% of European families, respectively, but in only a minority of patients from other ancestries. LRRC6 and CCDC103 were the most frequently mutated genes in South Asian families, affecting overall more than a third (20% and 16%, respectively) of families. CCDC39 and CCDC40 were the major two mutant genes affecting the Arab population, identified in 42% of Arab families (figure 1C). As detailed further below, these frequencies were due to a mixture of recurrent, presumed founder effect mutations, as well as mutations often unique to individual families.

PCD-causing mutation distribution in the multiancestry cohort is dominated by protein truncating and homozygous mutations. (A) Schematic of the DNAH5 mutations identified in this study, marked in yellow if previously reported. Recurrent mutations are boxed in bold. Conserved domains of the DNAH5 protein are indicated on the genomic structure. Variants are numbered according to (NM_001369.2) transcript. (B) Families with mutations in known PCD genes grouped based on their zygosity status showed that about 58% of mutations identified in this study were present in patients in a homozygous state. (C) Mutations classified according to their impact on the respective proteins showed that frameshift and nonsense mutations were the most prevalent (58%), with 15% splicing defects and 21% missense changes. Collectively, mutations predicted to have a protein truncating effect represent about 77% (frameshift, nonsense, splicing defects and CNV). (D) Mutations identified in biallelic state in autosomal gene or hemizygous state in X linked gene classified based on the guidelines from the American College of Medical Genetics showed that 82% of mutations were class 5 (clearly pathogenic), 8% class 4 (likely pathogenic) and 10% class 3 (uncertain significance). PCD, primary ciliary dyskinesia.
Expanded mutation spectrum in known PCD genes
A high proportion of families (74 of 128, 58%), from all ancestry groups, were found to carry homozygous variants (figure 2B). Surprisingly, one-third of Europeans families (20 of 60), considered largely non-consanguineous, carry homozygous variants in known PCD genes (online supplementary figure S3), highlighting possible unrecognised endogamy and relatedness. Biallelic heterozygous variants in autosomal genes caused disease in 31% of patients (39 families), and in 3 families hemizygous variants were identified in known X linked genes (PIH1D3, RPGR, OFD1).
Of the total 167 variants in known PCD genes detected in 128 families, the predominant variant types were predicted protein truncating mutations (73%) classified as frameshift (32%), nonsense (26%) and mutations affecting splicing (15%). Missense variants accounted for 21% of all variants. CNVs and inframe deletions or deletion/insertion mutations accounted for 6% of variants overall (figure 2C). Twelve single variants identified without a second mutation (‘one hit’ patients in online supplementary table S3) were not regarded as diagnostic, but among the 155 variants that diagnosed 116 families (excludes DNAH9 and CFAP300 alleles) 82% were pathogenic (class 5) and 8% were likely pathogenic (class 4). Class 3 variants of unknown clinical significance (VUS) represented only 10% of variants; these remain under some caution for providing a definitive diagnosis (figure 2D).
Marked differences in the frequency and spectrum of mutations in different ancestries
Across the cohort, in addition to the presence of many family-unique mutations, the prevalence of a number of recurrent mutations, presumed to reflect population bottleneck/founder effects, plays a major contribution to the different mutated genes affecting different PCD populations. We defined 14 recurrent mutations that collectively accounted for a large number of the PCD-causing variants. Some were ancestry-specific and others were present in multiple populations (table 2). In the 60 European families where causal alleles were defined, three recurrent DNAH5 mutations were identified to account for 13% (16 of 120) of European disease alleles, two previously reported as possible founder effects (c.10815delT; p.Pro3606Hisfs*22 and c.13458_13459insT; p.Asn4487fs*1)18 and a nonsense mutation (c.6261T>G; p.Tyr2087*) not previously reported (table 2, online supplementary table S3 and figure 2A). Three other previously reported mutations together accounted for another 13% (16 of 120) of European disease alleles: DNAI1 c.48+2 dupT; p.Ser17Valfs*12, RSPH1 c.275-2A>C; p.Gly92Alafs*10, and a recurrent homozygous DYX1C1 3.5 kb genomic deletion.19–22 DNAH11 mutations are also a major contributor to European PCD disease, but mostly as family-unique rather than recurrent alleles.
Ancestry-specific frequent mutations
In South Asian families, a previously described LRRC6 mutation (c.630delG; p.Trp210Cysfs*12) was the most frequent mutant allele, found in homozygous status in five South Asian families.23 A previously reported CCDC103 mutation was detected in homozygous state (c.383dupG; p.Pro129Serfs*25) in two unrelated South Asian families.24 Together, these two variants alone accounted for 28% (14 of 50) of all disease alleles in 25 South Asian families where causal alleles were defined (table 2 and online supplementary table S3). A known recurrent Arabic Bedouin RSPH9 mutation (c.801_803delGAA; p.Lys268del) was detected in homozygous state in two Arab families.19 Another possible Arabic homozygous founder mutation (c.1871_1872delTA; p.Ile624Lysfs*3) in CCDC39 was found in four Palestinian families. Together, these two variants accounted for 29% (12 of 48) of all disease alleles in the 24 Arabic families where causal alleles were defined (table 2 and online supplementary table S3). CCDC40 mutations also contribute to Arabic PCD disease, but in the form of family-unique rather than recurrent alleles.
Of other recurrent variants, the previously reported common South Asian CCDC103 missense mutation (c.461A>C; p.His154Pro) was detected mostly in South Asians, but also in European and other ancestries.25 We also identified in different ancestries two recurrent CCDC40 mutations, one previously reported (c.248delC; p.Ala83Valfs*84)20 21 and one novel (c.2824_2825insCTGT; p.Arg942Thrfs*57), in addition to one novel DNAH11 mutation (c.13494_13500del; p.Ser4498Argfs*15).
Targeted NGS reveals synonymous variants predicted to affect splicing as a cause of PCD
We identified two synonymous coding region variants not predicted to change the encoded protein’s amino acid sequence, but predicted instead to affect splicing. One, in an Arab family (PCD-G086) with cilia microtubular disorganisation and inner dynein arm (IDA) loss, was a CCDC40 homozygous variant (c.48A>G; p.Gly16Gly) that correctly segregated within the extended family (online supplementary figures S4 and S5). The other, in a European family (PCD-G093) with cilia ODA loss, was a DNAH5 synonymous mutation (c.5157C>T; p.Phe1719Phe) that was combined with a missense variant (c.10815T>G; p.Asp3605Glu) (online supplementary figures S6 and S7). While it is possible these synonymous variants may affect splicing, they are currently class 3 VUS (online supplementary table S3) and cannot be reclassified to pathogenic or likely pathogenic without further work that provides direct observation of their presumed splicing effects.
Targeted NGS is a powerful tool for diagnosis and characterisation of patients with PCD
TEM analysis detected ultrastructural defects in 97 of 134 (72%) families. The other 37 had either normal TEM (eg, associated with DNAH11 and HYDIN defects26 27) or a minority of inconclusive TEM results (table 1). The most common ultrastructural defect was ODA loss (in 45%, 61 families), either alone (in 23%, 31 families) or combined with IDA loss (in 22%, 30 families). Other defects included microtubular disorganisation with or without IDA loss (12%), central microtubular complex defects (6%), predominant isolated IDA loss (4%) or a lack of cilia (5%) (table 1).
We confirmed a strong correlation across the entire cohort between gene defect and expected ultrastructural defect, in agreement with the PCD literature (table 1 and online supplementary table S1).2 Hence, TEM defects can be valuable for interpretation of genetic test results; however, the study also showed that they are not always required. For the 27 of 161 (17%) families without TEM data, still with strong clinical suspicion, 18 had biallelic variants in known PCD genes; hence, a high proportion (67%) were confidently solved by genetics without TEM information (online supplementary figure S8). As a cautionary note, for a small number of patients (n=6, asterisks in online supplementary table S3) without a recorded TEM defect confirming of their PCD status, they also carry homozygous or biallelic heterozygous variants that are rare and in the known PCD genes, but are class 3 VUS of uncertain significance. For example PCD-G013 is biallelic heterozygous for two DNAH5 missense changes both of unknown significance (not previously reported). In these cases, three have variants in the HYDIN and DNAH11 genes associated with normal TEM (PCD-G104, PCD-G017, PCD-G021), but for the others the TEM could be reviewed and repeated.
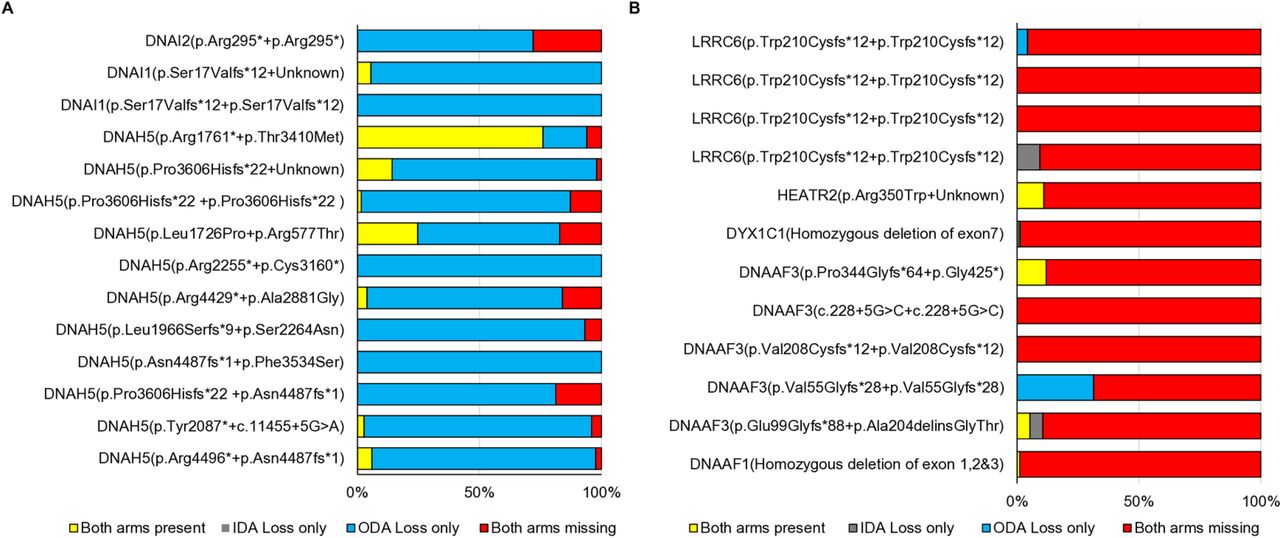
To further test the power of genetic testing in the diagnostic workflow of PCD, we looked in detail at the correlation of specific PCD gene mutations with ciliary ultrastructural defects determined by TEM at a single diagnostic centre. We found that mutations in the ODA gene DNAH5 were associated with (1) clear-cut ODA loss as expected, but could sometimes also be recorded to have (2) combined loss of IDA+ODA or (3) inconclusive TEM analysis (figure 3A, table 1). A similar classification was possible in individuals with dynein assembly gene mutations (LRRC6, HEATR2, DYX1C1, DNAAF3 or DNAAF1), where combined IDA+ODA loss is expected (figure 3B, table 1). By looking at the TEM data in the context of the genetic mutation, we were able to define a distinct pattern and difference between these two gene function categories, since dynein assembly mutations led to combined IDA+ODA loss in most cilia cross sections, contrasting with DNAH5 mutations causing mainly ODA loss. Therefore, genetic data allow these two categories of genetically diagnosed patients to be distinguished (figure 3).

{kind=link}
{kind=link}
{kind=link}
Genetics can better characterise patients with PCD and overcome other diagnostic testing inconsistencies. For a selected set of patients, the percentage of cilia cross sections showing a loss of either or both the inner and outer dynein arms was recorded in the routine TEM diagnostic setting at Royal Brompton Hospital with reference to the underlying genetic defects. (A) Patients carrying biallelic mutations in ODA components showed mainly an isolated loss of ODA. Combined IDA+ODA (inner and outer dynein arm) loss was also noted. Interestingly quite variable numbers of cross sections showed a normal ultrastructure of the cilia. (B) Patients carrying biallelic mutations in dynein assembly genes showed a combined loss of both arms in the majority of cross sections examined. Variable isolated arm loss was also reported. ODA, outer dynein arm; PCD, primary ciliary dyskinesia; TEM, transmission electron microscopy.
Discussion
There is high underlying disease heterogeneity and no gold standard test available yet to exclude PCD, so a combination of tests interpreted in the light of clinical symptoms tends to be used for diagnosis. This increasingly includes genetic analysis.8 Here, the utility of genetic screening was evaluated in a large cohort of 161 unrelated PCD families from various ancestries including European, Arab and South Asian, by NGS screening with additional CNV analysis of the known PCD genes and a panel of other candidate genes. This gave a high yield of a confirmed or highly suggested PCD diagnosis in 75% of families. A further 7% of families with single heterozygous variants in known PCD genes that looked likely causal may likely carry a second mutation that NGS was not able to detect, for example a deep intronic variant.
The identification of clear-cut ciliary ultrastructural defects by TEM analysis remains a confirmatory step in the PCD diagnostic workflow, although failure to identify TEM defects does not exclude PCD.8 Here, we identified mutations in known PCD genes in 81% of patients with normal or inconclusive TEM findings, implying significant potential for incorporating genetics earlier within the diagnostic pipeline, as previously discussed.27 We could also diagnose 67% of patients with a strong history where TEM was not available, as well as other difficult cases, for example, CCDC103 p.His154Pro mutations, where other tests often give equivocal results.25
Our diagnostic output is higher than most previous NGS targeted panel screens in PCD,22 28–30 similar to the 76% diagnostic success achieved from whole exome sequencing (WES) and targeted CNV analysis in 52 individuals.31 32 The limitations include the unknown genes that are absent from the panel, the incomplete genetic diagnosis when variants of uncertain significance or single heterozygous variants in PCD genes are detected, technical issues affecting sequence coverage depth, the known bioinformatics challenges to identify CNVs,33 and a well-known problem with identification of HYDIN mutations, due to the HYDIN2 copy gene.26
This study expands the genetic landscape and mutation spectrum of PCD by identifying 61 previously reported and 88 previously unrecognised variants; hence, 57% of all the variants classified here as likely pathogenic are novel (online supplementary table S3). Most were protein truncating mutations, predicted to be loss-of-function alleles, consistent with previous reports. Synonymous mutations are not commonly reported in PCD, but we identified two predicted to result in alteration of splicing, raising the importance of looking for potential synonymous variants in unsolved cases.
In agreement with previous studies, most of the identified PCD variants were private1 8; however, several were detected in more than one unrelated family that tended to be more frequent in certain populations. Interestingly, one-third of mutations in European families were homozygous despite the low recorded European consanguinity rate, with only one European family reporting consanguineous marriage. In 14 European families, their identified mutations were reported before in the literature, suggesting they may reflect European founder effects. Overall, we found that DNAH5 is the most commonly mutated PCD gene in agreement with other studies,34 but this was not the case in all ancestries. We identified DNAH5 mutations in 37% of European families, representing the most common mutant gene in Europeans. In contrast, LRRC6 and CCDC103 mutations were more prevalent in South Asian families, among whom we found only one family with DNAH5 mutations. In Arab families, DNAH5 mutations were identified in only two families, with CCDC39, CCDC40 and RSPH9 mutations much more prevalent.
The study has therefore uncovered a striking population-based genetic stratification underlying PCD. It highlights the impact of ancestry on the genetics of PCD and the importance of including patients from various ancestries to elucidate the full genetic landscape of PCD. This information is diagnostically relevant, as it could be used for improved, smaller/cheaper carrier screening panels targeting certain populations and preliminary allelic-specific genetic diagnosis by Sanger sequencing, especially in countries where NGS facilities are not widely available. The clinical relevance of genetic disease stratification remains poorly understood, but more studies are emerging with PCD genotype–phenotype correlations which can increasingly impact on disease management.17 ,4 35–37
Although we could confirm good correlation between genotype and cilia ultrastructural phenotype, some differences in the TEM analysis results were evident even with mutations in the same gene. DNAH5 mutations were associated with ODA loss in the majority of cases but also with some cases of inconclusive TEM results, possibly due to difficulties in evaluating IDA by TEM,3 8 as well as with occasional recording of combined IDA+ODA loss that is more often linked to dynein assembly gene mutations. Quantification of the percentage of arm loss arising from DNAH5 mutations compared with mutations in dynein assembly genes showed that augmenting TEM data with genetics could clearly distinguish these two groups, DNAH5 mutations being more highly linked to ODA loss and dynein assembly mutations more highly to combined IDA and ODA loss.
In conclusion, targeted multigene panel sequencing is a cost-effective, time-efficient single test which in this studyconfidently diagnosed at least 75% of PCD cases. It improves the diagnostic workflow outcome, confirming PCD in patients with inconclusive TEM results and helping in the diagnosis of patients where TEM analysis is not available. The sensitivity (diagnostic yield) of gene tests for PCD will continue to increase with gene discovery progress. CFAP300, DNAH9, GAS2L2, LRRC56, MNS1, DNAH1 and DNAH6 are all genes that have become associated with PCD or confirmed as PCD genes in the interim study period.16 17 38–42 Despite the current incomplete PCD gene list, this strongly supports the importance of including genetics into the diagnostic pathway where it can play a key role, overcoming the pitfalls of other diagnostic measures. This may be particularly relevant in countries where access to other specialised PCD tests is not available. Major impact genes and recurrent mutations have emerged in this study, in addition to a notable impact of ancestry on the genetic variability of PCD, which has implications for the improved stratification of patients with PCD to help facilitate better targeting of diagnostics and disease management.
Acknowledgments
We are very grateful to the families who participated in this study and thank the UK PCD Family Support Group for their continued support. We thank all laboratory and clinical members of the national PCD Diagnostic Service at UHS, RBH and LRI who have contributed to the PCD diagnostic testing. We thank Andrew Jarman, Cecilia Lo and Sudipto Roy for prepublication candidate PCD gene lists. We thank Dr Lisa Robertson, Clinical Genetics Service, Aberdeen Royal Infirmary, NHS Grampian, for her clinical involvement.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MahmoudFassad
Correction notice This article has been corrected since it was published Online First. Affiliation 22 has been corrected.
Contributors MRF, MP, TC, JH, LJ, DJM-R, CMW, HM and HMM performed the genetic and bioinformatic analysis and analysed the genetics data. AS, MD, AVR, SO, CJ, PG, RH, AR, JT, AP and SL were involved in generating and analysing clinical functional tests. AS, MD, AVR, PG, AR and AP performed cilia ultrastructural analysis. MRF, PA, EM, PC, COC, RW, SC, WW, HM, WS, LP, CC, ML, EMKC, PK, NR, NF, JSL, CH and HMM ascertained patients and acquired patient data and samples. HMM conceived and supervised the study and data interpretation. MRF interpreted the genetics data, developed the manuscript draft and generated the figures. MRF and HMM wrote the manuscript with critical review input from AS, TC, CMW, JSL and CH. All authors reviewed and approved the final manuscript.
Funding The national PCD Diagnostic Service is funded by NHS England. Research in Southampton is supported by NIHR Southampton Respiratory Biomedical Research Unit, NIHR Wellcome Trust Clinical Research Facility and the AAIR Charity (Reg No 1129698). Funding for this study was provided by Action Medical Research (GN2101; HMM) and Great Ormond Street Children’s Charity grant (V4515; HMM) and Leadership awards (V1299, V2217; HMM). We acknowledge support from the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London (Doctoral Trainee Support Award; MRF). MRF is supported by the British Council Newton-Mosharafa Fund and the Ministry of Higher Education in Egypt. Work by AS is an independent research funded by a postdoctoral research fellowship from the National Institute for Health Research and Health Education England. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. The authors participate in and acknowledge financial support from the COST Action BEAT-PCD: Better Evidence to Advance Therapeutic options for PCD network (BM1407), in particular two STSM grants awarded to MRF.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Patient recruitment took place with informed, age-appropriate consent as approved by the London-Bloomsbury Research Ethics Committee (08/H0713/82) and the committees of collaborating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable request.
Author note The authors are members of COST Action BEAT-PCD (BM1407).