Pulmonary hypertension: diagnosis and management
BMJ 2013; 346 doi: https://doi.org/10.1136/bmj.f2028 (Published 16 April 2013) Cite this as: BMJ 2013;346:f2028
- David G Kiely, consultant respiratory physician1,
- Charlie A Elliot, consultant respiratory physician1,
- Ian Sabroe, consultant respiratory physician12,
- Robin Condliffe, consultant respiratory physician 1
- 1National Pulmonary Hypertension Service (Sheffield), Pulmonary Vascular Disease Unit, Royal Hallamshire Hospital, Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield S10 2JF, UK
- 2Academic Unit of Respiratory Medicine, Department of Infection and Immunity, Faculty of Medicine, Dentistry and Health, Medical School, University of Sheffield, Sheffield, UK
- Correspondence to: D G Kiely david.kiely{at}sth.nhs.uk
Summary points
Pulmonary hypertension has many causes so prognoses and treatments vary
The condition is diagnosed by systematically evaluating the breathless patient and screening patients at high risk
Patients at high risk of severe and treatable pulmonary hypertension include those with systemic sclerosis, portal hypertension, congenital heart disease, and previous pulmonary embolism
Specialist centres provide access to tailored investigative and treatment pathways and support networks for patients
Patients with severe pulmonary hypertension can deteriorate rapidly—do not delay referral to perform specialist investigations
Only selected patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension benefit from interventions directed at pulmonary vasculature; for most patients treatment is aimed at the underlying condition
Pulmonary hypertension was previously considered a rare untreatable condition. The past two decades have seen major changes in our understanding of the spectrum of disease affecting the pulmonary circulation. More than 30 randomised controlled trials (RCTs) of treatments have been performed, and surgery for patients with chronic thromboembolic pulmonary hypertension has been developed. Advances in imaging have enabled more detailed patient assessment, but pulmonary hypertension continues to be a life shortening condition, and there is often a delay of around two years from onset of symptoms to diagnosis.1 This article focuses on adult pulmonary hypertension in primary and secondary care, its diagnosis, and management.
Sources and selection criteria
This review is based on our personal experience, personal archives of references, and a PubMed search using terms including pulmonary hypertension, chronic thromboembolic disease, and prognosis. We consulted international guidelines from the European Society of Cardiology, European Respiratory Society, and American College of Chest Physicians.
What is pulmonary hypertension and how common is it?
Pulmonary hypertension is defined at cardiac catheterisation as a mean pulmonary artery pressure of 25 mm Hg or more. The initial clinical classification in 1973 arose from a World Health Organization sponsored international meeting after an epidemic related to use of the appetite suppressant, aminorex fumarate.2 It defined primary pulmonary hypertension (now termed idiopathic pulmonary arterial hypertension) as being characterised by vasculopathy of the pulmonary arteries. Better understanding of disease mechanisms led to subsequent classification of conditions with shared clinical and pathophysiological characteristics:
Group 1: Pulmonary arterial hypertension (PAH), which can be idiopathic (IPAH) or associated with other conditions, notably systemic sclerosis and congenital heart disease
Group 2: Pulmonary hypertension owing to left heart disease (PH-LHD)
Group 3: Pulmonary hypertension owing to lung disease or hypoxia (PH-Lung), or both
Group 4: Chronic thromboembolic pulmonary hypertension (CTEPH)
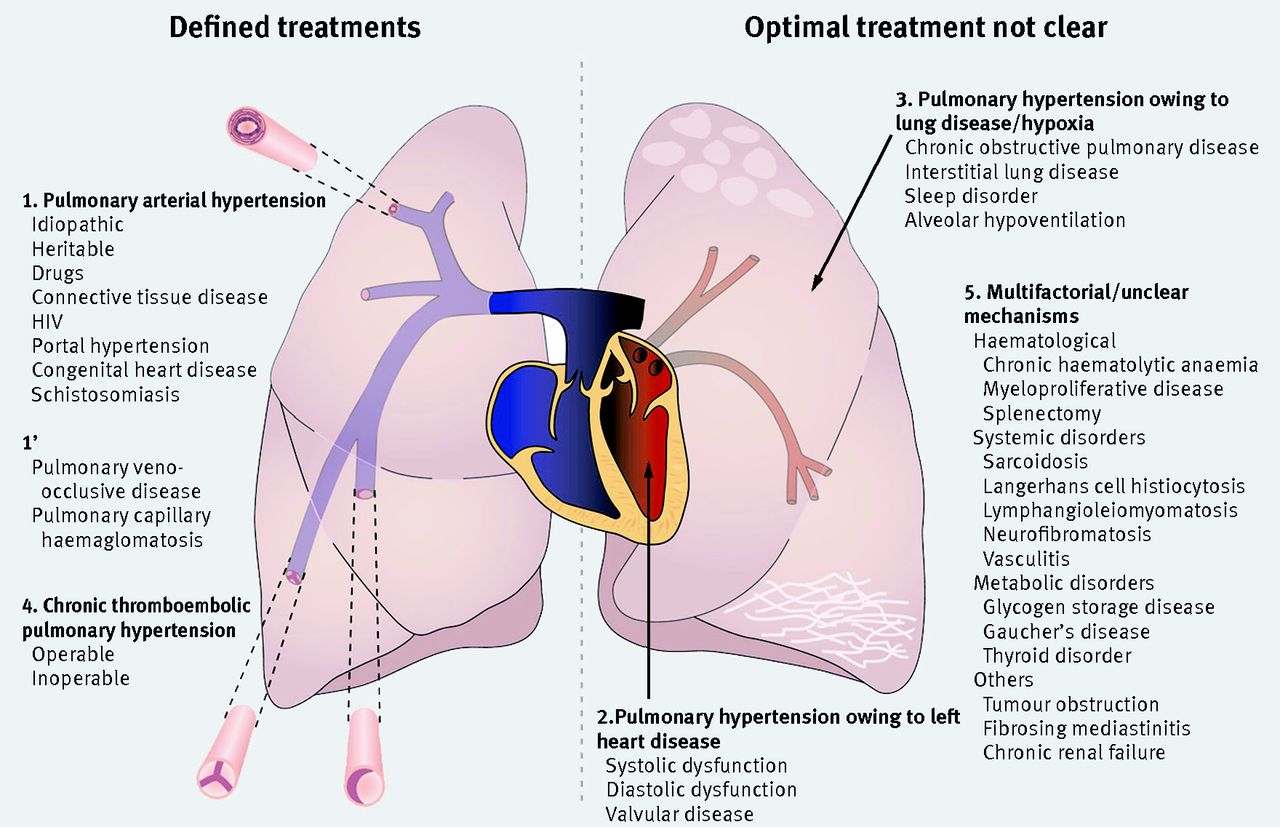
Fig 1 Classification of adult pulmonary hypertension. The condition can be caused by narrowing of the pulmonary arterial tree as a result of vasculopathy (group 1) or webs and stenoses of chronic thromboembolic disease (group 4). Optimal treatments are generally defined for groups 1 and 4. More commonly, it is associated with left heart disease (group 2) or lung disease (group 3), and optimal treatments are unclear. Several conditions associated with pulmonary hypertension have multifactorial or unclear mechanisms (group 5). *Chronic haemolytic anaemia will probably be in group 5 in next classification update
{kind=link}
Many mechanisms can lead to elevation of pulmonary pressures. In PAH, progressive narrowing of the pulmonary arterial bed results from an imbalance of vasoactive mediators, including prostacyclin, nitric oxide, and endothelin-1. This leads to an increased right ventricular afterload, right heart failure, and premature death. Between 11% and 40% of patients with IPAH and 70% of patients with a family history of PAH carry a mutation in the gene encoding bone morphogenetic receptor-2 (BMPR2); however, penetrance is low—carriers have a 20% lifetime risk of developing pulmonary hypertension.4 5 Therefore, “multiple hits” are probably needed for the development of PAH. In PH-LHD, raised left atrial pressures result in secondary elevation of pulmonary pressure. In PH-Lung, raised pulmonary arterial pressures result from mechanisms such as vascular destruction and hypoxic vasoconstriction. In CTEPH, mechanical obstruction of the pulmonary vascular bed is the primary process.
Incidences are estimated to be 1-3.3 per million per year for IPAH and 1.75-3.7 per million per year for CTEPH; the prevalence of PAH is estimated at 15-52 per million.6 7 8 9 10 Pulmonary hypertension is more common in severe respiratory and cardiac disease, occurring in 18-50% of patients assessed for transplantation or lung volume reduction surgery, and in 7-83% of those with diastolic heart failure.11 12 13 14
Who gets pulmonary hypertension and why is classification important?
IPAH is rare and primary care doctors may never encounter a case. By contrast, the prevalence of PAH is high in certain patient groups, such as those with systemic sclerosis (9%), portal hypertension (2-6%), congenital heart disease (5-10%), and HIV (0.5%).15 16 17 18 19 20 Between 0.5% and 4% of patients develop CTEPH after acute pulmonary embolism; patients at increased risk include those presenting with large, recurrent, or unprovoked clots.21 22 Classification is crucial in determining treatment and prognosis.9 Specific treatments exist for PAH and CTEPH. By contrast, in PH-LHD and PH-Lung, treatment is best directed at the underlying condition, and currently PAH specific treatments are not recommended. Distinguishing between IPAH and PH-LHD due to diastolic dysfunction in the setting of normal left ventricular systolic function may be challenging (table 1⇓).
Pulmonary hypertension in patients with normal left ventricular systolic function: features useful in discriminating between idiopathic pulmonary arterial hypertension and pulmonary hypertension caused by left ventricular diastolic dysfunction
Median survival in untreated IPAH is 2.8 years, but more recent registries have observed overall median survival of at least five years.9 23 In the past, the mean age at diagnosis in IPAH was 35 years; current five year survival in this younger age group is now about 75%.10 PAH associated with systemic sclerosis carries a worse prognosis than IPAH, whereas the opposite is true for congenital heart disease.9 The development of pulmonary hypertension in cardiac and respiratory disease has a negative impact on prognosis, which depends on the underlying cause and severity of pulmonary hypertension.
What are the symptoms and signs of pulmonary hypertension?
PAH most commonly presents with progressive breathlessness.24 As right ventricular dysfunction develops patients can experience exertional dizziness and syncope. Oedema and ascites occur late in the disease. Anginal chest pain may occur due to exertional myocardial hypoperfusion in the setting of right ventricular hypertrophy, left main coronary artery compression by the pulmonary artery, or ischaemic heart disease.25 Patients are prone to tachyarrythmias, most commonly atrial flutter, which can cause sudden decompensation.26 Haemoptysis, although uncommon, can occur in Eisenmenger’s syndrome and CTEPH, where bronchial arteries may be enlarged. Signs suggestive of pulmonary hypertension include a loud pulmonary second heart sound, systolic murmur of tricuspid regurgitation, raised jugular venous pressure, peripheral oedema and ascites, but these signs may be subtle or absent in early disease. Look for signs of associated conditions (such as connective tissue disease or liver disease).
When should I suspect pulmonary hypertension?
Pulmonary hypertension is diagnosed by systematically evaluating the breathless patient and screening high risk groups. Unexplained progressive exertional breathlessness in the absence of symptoms or signs of respiratory or left heart disease is suggestive. Screening protocols exist for high risk groups. Rheumatology centres screen patients with systemic sclerosis annually, and potential liver transplant recipients are routinely investigated for portopulmonary hypertension. Persistent breathlessness after pulmonary embolism should alert the doctor to the possibility of CTEPH.
What investigations should I perform if pulmonary hypertension is suspected?
Electrocardiography, chest radiography, and pulmonary function tests may identify an alternative cause of breathlessness (fig 2⇓). Evidence of right heart strain on electrocardiography and prominent pulmonary arteries or cardiomegaly on chest radiography were seen in 80-90% of cases in a large IPAH registry.24 In clinical practice, however, a normal electrocardiograph and chest radiograph cannot exclude the diagnosis. A reduction in gas transfer (lung capacity to “transfer” an inhaled gas into the blood) and mildly deranged spirometry both occur in IPAH.27 If pulmonary hypertension is still suspected, echocardiography should be performed.28 Figure 3⇓ describes the estimation and interpretation of systolic pulmonary artery pressure, together with other echocardiographic features suggesting the presence of pulmonary hypertension. Importantly, pulmonary hypertension is defined by mean pulmonary artery pressure (measured at right heart catheterisation) not systolic pulmonary artery pressure (estimated by echocardiography). In addition, systolic estimates are less accurate in respiratory disease and during intercurrent illness.14

Fig 2 Diagnostic pathway for patients with suspected pulmonary hypertension
{kind=link}

Fig 3 Echocardiographic assessment of pulmonary hypertension. RA=right atrium; RV=right ventricle; LA=left atrium; LV=left ventricle
{kind=link}
How is pulmonary hypertension assessed in a specialist unit?
If initial investigations suggest that pulmonary hypertension is possible, further tests are needed to confirm the diagnosis and to establish cause and severity. In the United Kingdom, a network of specialist centres was commissioned in 2001 to standardise and improve care (box 1). If severe disease is suspected, do not delay referral to a specialist centre, although imaging to exclude thromboembolic disease may be considered. If the degree of pulmonary hypertension seems to be disproportionate to the underlying cardiorespiratory disease, or if diagnostic doubt exists, discussion with a specialist centre is recommended. In these groups it is also important to exclude coexisting thromboembolic disease as a cause of pulmonary hypertension. Accurate classification and assessment of disease severity (fig 2) requires integration of clinical, physiological, and radiological data. This is central to further management (fig 4⇓).
Box 1 UK designated pulmonary hypertension centres for adults and children
Nationwide
UK national centres: www.pulmonaryhypertensioncentres.co.uk
Glasgow
Golden Jubilee Hospital: www.spvu.co.uk
Newcastle
Freeman Hospital: www.newcastle-hospitals.org.uk
Sheffield
Royal Hallamshire Hospital: www.thoracic.group.shef.ac.uk
Cambridge
Papworth Hospital: www.papworthhospital.nhs.uk
London
Royal Brompton Hospital: www.rbht.nhs.uk
Hammersmith Hospital: www.pulmonary-hypertension.org.uk
Royal Free Hospital: www.royalfree.nhs.uk
Great Ormond Street (children): www.gosh.nhs.uk

Fig 4 Overview of patient pathway at a specialist centre
{kind=link}
Blood, exercise testing, and overnight oximetry
Immunological and HIV testing may identify a cause of pulmonary hypertension. Brain natriuretic peptide (BNP) and N-terminal-pro-BNP have prognostic importance. Lower levels at baseline and improvement during follow-up are associated with better outcomes.29 30 The six minute walk test has been extensively evaluated. It provides functional and prognostic information and can be used to monitor treatment response.31 32 Cardiopulmonary exercise testing in selected patients gives more detailed physiological and prognostic information.33 Overnight oximetry is performed if sleep disordered breathing is a suspected cause.
Imaging
Isotope perfusion lung scanning has high sensitivity for CTEPH and a normal scan excludes the diagnosis, whereas increased renal isotope uptake may identify right to left shunts and explain hypoxaemia.34 35 High resolution computed tomography and computed tomography pulmonary angiography visualise the pulmonary arterial tree and provide a wealth of additional diagnostic information (fig 5⇓). Direct features of chronic thromboembolic disease include occlusions, stenoses, and intraluminal webs (fig 1), whereas indirect signs include a mosaic perfusion pattern. These signs can be subtle and may be missed by a non-specialist radiologist. Magnetic resonance imaging provides a radiation-free method of quantitatively assessing cardiac structure and function, prognosis, and response to treatment.36 37 Magnetic resonance pulmonary angiography can supplement computed tomography pulmonary angiography in the assessment of CTEPH operability, and magnetic resonance perfusion imaging is as sensitive as isotope perfusion lung scanning.38 39 Pulmonary angiography is not commonly used in the UK to select CTEPH patients for pulmonary endarterectomy, but is used widely in other centres throughout the world to assess for surgical accessibility.

Fig 5 Computed tomography assessment of pulmonary hypertension. RA=right atrium; RV=right ventricle, LA=left atrium; LV=left ventricle; IVS, interventricular septum; PA, pulmonary artery; Ao, aorta; CTEPH=chronic thromboembolic pulmonary hypertension
{kind=link}
Right heart catheterisation
If non-invasive investigations support a diagnosis of pulmonary hypertension, right heart catheterisation is needed to confirm the diagnosis by directly measuring pulmonary pressure. It also allows measurement of cardiac output and estimation of left atrial pressure using pulmonary arterial wedge pressure. Haemodynamic parameters (such as right atrial pressure, cardiac index, and pulmonary vascular resistance) and pulmonary artery saturations have important prognostic value. Raised pulmonary arterial wedge pressure is suggestive of PH-LHD. Changes in oxygen saturations between right sided cardiac chambers may suggest an intracardiac shunt. Vasoreactivity testing, usually using inhaled nitric oxide, can identify patients with IPAH who may respond to long term high dose calcium channel blockers. A positive response is defined as a reduction in mean pulmonary artery pressure of at least 10 mm Hg, to less than 40 mm Hg, without a fall in cardiac output.40
What treatments are available for pulmonary hypertension?
Supportive treatment
Patients with CTEPH require anticoagulants because of the central role of thromboembolic disease. Although anticoagulation is recommended in IPAH, its role in PAH associated with other conditions is less clear.41 42 Diuretics are often needed to treat heart failure. Digoxin and long term oxygen therapy may be considered, but there is no evidence from RCTs on their use. Annual flu vaccination is recommended.43 Pregnancy is poorly tolerated despite changing practices,44 and a recent systematic review found that maternal mortality remains high (17-33%).45 Women are counselled about the high risks and are offered effective contraception; termination is recommended if pregnancy occurs.43 Progesterone-only contraceptives—oral desogestrel, intramuscular medroxyprogesterone, and etonogestrel implants—avoid the prothrombotic effects of combined oral contraceptives. The levonorgestrel releasing coil is effective, but poor tolerance of potential vasovagal episodes mandates insertion in hospital. Emergency hormonal contraception is safe after unprotected intercourse. Non-obstetric surgical intervention also poses risks and its indication requires careful evaluation.
Specific medical treatment (table 2⇓)
Licensed drugs for treating pulmonary hypertension
High dose calcium channel blockade
About 10% of patients with IPAH have a positive response to vasoreactivity testing at right heart catheterisation. Observational studies suggest that half of these patients exhibit long term clinical responses to high dose calcium channel blockade (such as diltiazem titrated to 480-720 mg/day or nifedipine titrated to 60-120 mg/day), with near normalisation of prognosis.46 47 No evidence exists for this treatment in other forms of PAH, and because of the potential negative inotropic effect, treatment should not be started without a positive acute vasoreactive test.
Prostanoids
Prostacyclin is a vasodilator with antiproliferative effects. Infused continuously through an indwelling central line, it was the first PAH specific drug shown to be efficacious in a small RCT, demonstrating improvements in exercise capacity, haemodynamics, and, uniquely, survival.48 It has a half life of about six minutes, is unstable at room temperature, and intravenous administration has an associated risk of line infection. Alternative routes of administration were therefore developed. A larger RCT found that nebulised iloprost (half life about 30 minutes) improved exercise capacity and haemodynamics when given seven times a day.49 Because of its superior stability, iloprost is also given intravenously in some centres. Treprostinil (half life about four hours) was shown, in a very large RCT, to significantly improve exercise capacity when given by continuous subcutaneous infusion.50 Further RCTs also showed significant improvements in exercise capacity when it was given by nebulised and intravenous routes.51 52 Oral prostanoids have also been assessed with large RCTs, but results have generally been disappointing, and studies are ongoing.53 54 55 56
Endothelin receptor antagonists
Endothelin is a potent vasoconstrictor of vascular smooth muscle. Two oral endothelin receptor antagonists are currently licensed, bosentan and ambrisentan, on the basis of well conducted RCTs showing improved exercise capacity and time to clinical worsening.57 58 Bosentan causes reversible abnormalities in liver function tests in 5-10% of patients, so monthly monitoring is needed.59
Phophodiesterase-5 inhibitors
Nitric oxide acts via cyclic GMP to increase pulmonary vascular relaxation and reduce cellular proliferation and is degraded by phophodiesterase-5. Two oral phophodiesterase-5 inhibitors (sildenafil and tadalafil) improved exercise and functional capacity in large well conducted RCTs.60 61 These drugs have important interactions with nitrates, so concomitant use is contraindicated.
Treatment approach
Guidelines suggest starting oral treatment for patients with PAH in WHO functional classes II and III, and considering parenteral prostanoids in suitable patients in class IV or those with poor prognostic markers in class III (box 2).43 If response to treatment is suboptimal, switching or adding further drugs is suggested.43 Sequential addition of drugs to achieve predefined treatment goals in six minute walk distance, maximal oxygen capacity, and peak systolic blood pressure was associated with improved outcome compared with patients whose treatment had been escalated in a non-standardised manner.62 Several RCTs of combination therapy are ongoing. Assessment of the patient’s ability to use complex prostanoid therapy and adherence to treatment require the support of experienced healthcare professionals.
Box 2 WHO functional classes
Class I: Patients with pulmonary hypertension but in whom ordinary physical activity does not cause undue symptoms
Class II: Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest but ordinary physical activity causes undue dyspnoea or fatigue, chest pain, or near syncope
Class III: Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest but less than ordinary activity causes undue dyspnoea or fatigue, chest pain, or near syncope
Class IV: Patients with pulmonary hypertension who cannot carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnoea or fatigue may be present at rest. Discomfort is increased by any physical activity
Chronic thromboembolic pulmonary hypertension
About two thirds of patients with CTEPH are candidates for pulmonary endarterectomy. This operation offers the best prospect of substantial improvement in symptoms, haemodynamics, and long term survival, as shown by a recent UK observational study.8 Surgical eligibility criteria are not clearly established, and suitable candidates are identified after assessment at specialist surgical centres (Papworth Hospital in the UK). Perioperative mortality in large volume centres is now less than 5%. In inoperable cases PAH specific drugs are often used, although evidence is suboptimal. Do not delay referral for surgery for trials of medical therapy, although such treatment can be used to support patients with severe CTEPH as a “bridge” to surgery.
Other non-medical treatments
Lung transplantation (usually double lung), which has overall five year survival of around 50%, is indicated in severe PAH or inoperable CTEPH if medical management fails.63 Atrial septostomy decompresses the pressure loaded heart and may be useful in patients with severe PAH who have intractable syncope but not severe hypoxaemia, or as a bridge to transplantation.64 Supervised exercise training improves exercise capacity, but effects on haemodynamics and prognosis are unclear.65
What challenges are on the horizon?
Despite progress in the management of PAH, it remains a life shortening condition. Understanding disease mechanisms and developing new treatments is a focus of international collaboration. Recent data have shown efficacy for drugs targeting existing pathways, such as macitentan (endothelin receptor antagonist)66 and riociguat (nitric oxide pathway via soluble guanylate cyclase stimulation).67 68 Selexipag,69 a prostacyclin receptor agonist, is also under investigation. Despite promising effects on pulmonary haemodynamics the tyrosine kinase inhibitor, imatinib, has recently been withdrawn from licensing application, partly because of safety concerns.70 Studies with stem cell therapy are ongoing.71 The role of mechanical treatments, such as cardiac resynchronisation and right ventricular assist devices, in PAH are of interest.72 73 The optimal treatment of PH-LHD and PH-Lung is unclear and drugs are being studied in phase III trials. Pulmonary artery balloon angioplasty is being investigated in patients with CTEPH who are not amenable to pulmonary endarterectomy.74 Future availability of generic oral drugs for pulmonary hypertension may lead to inappropriate prescribing in PH-LHD and PH-Lung. It may also increase late referral to specialist centres and delays in escalation of treatment.
Improved diagnostics and treatments provide increasingly complex choices for patients and healthcare professionals in this rare illness. The delivery of care through specialist centres is increasingly advocated worldwide as a cost effective way to deliver high quality care.43 Education on disease trajectory, provision of psychological support, and access to social and end of life care are championed by patients and their representative organisations, and they are key to improving the quality of life for patients with pulmonary hypertension.75
Tips for non-specialists
Consider pulmonary hypertension as a cause of progressive unexplained breathlessness
Echocardiography is the most useful non-invasive test for investigating pulmonary hypertension but estimates systolic, not mean, pulmonary artery pressure
Computed tomography pulmonary angiography is useful for assessing suspected pulmonary hypertension and also allows estimation of cardiac chamber size
A large left atrium suggests that pulmonary hypertension is more likely to be caused by left heart disease
Pulmonary hypertension should not be empirically treated until a clear diagnosis is established
If a patient has suspected severe pulmonary hypertension remember that the differential diagnosis is wide and outlook has improved considerably over the past 10 years
Additional educational resources
Resources for healthcare professionals
European Society of Cardiology (www.escardio.org/guidelines)—Guidelines for the management of pulmonary hypertension
Pulmonary Hypertension Association UK (www.paheducation.co.uk)—elearning course for healthcare professionals developed to educate non-specialist healthcare professionals including general practitioners, nurses, and clinicians about pulmonary hypertension and its management
Pulmonary Hypertension Online University (www.phaonlineuniv.org)—Specialist internet resource for continuing medical education
Pulmonary Vascular Research Institute (www.pvri.info)—Independent medical research organisation devoted to increasing the awareness and knowledge of pulmonary vascular diseases and to facilitating advances in the treatment of affected people worldwide
Resources for patients
UK Pulmonary Hypertension Patients Association (www.phassociation.uk.com)—Supports adults and children with pulmonary hypertension and their families
PHA Europe (www.phaeurope.org)—Information for patients with pulmonary hypertension as well as their family and friends
UK Raynaud’s and Scleroderma Association (www.raynauds.org.uk)—Supports patients with Raynaud’s syndrome and scleroderma
The Sommerville Foundation (www.thesf.org.uk)—Supports young people and adults born with a heart condition (congenital)
Notes
Cite this as: BMJ 2013;346: f2028
Footnotes
We thank Heather Allen for her help with fig 1.
Contributors: DGK, CAE, IS, and RC conceived and designed the article and approved the final manuscript. DGK is guarantor.
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organisation for the submitted work; DGK has received speaker fees, attended advisory boards, and received funding to attend meetings from Actelion, Bayer, Pfizer, United Therapeutics, Lily, and GSK; CAE has received speaker fees and attended advisory boards for Actelion, Bayer, and GSK; IS has received travel funding from Actelion and GSK; RC has received speaker fees from Actelion and GSK and has attended advisory boards for Actelion, Bayer, and GSK. The department has received funding for research and education through unrestricted grants from Actelion, Bayer, and Pfizer.
Provenance and peer review: Commissioned; externally peer reviewed.