Article Text

Statistics from Altmetric.com
- AATD, alpha1-antitrypsin deficiency
- COPD, chronic obstructive pulmonary disease
- OR, odds ratio
- 95% CI, 95% confidence interval
Alpha1-antitrypsin deficiency (AATD) is a genetic condition associated with an increased risk of developing chronic obstructive pulmonary disease (COPD) early in life and, to a lesser extent, liver disease.1 Significant advances have been made during the last decades in understanding its epidemiology and it has been recently suggested that AATD is one of the commonest inherited disorders not only in Caucasians but also among other ethnic groups worldwide.2,3
Although AAT is a highly pleomorphic glycoprotein, with approximately 100 variants having being identified,4 two major deficient variants, namely Z and S, account for most cases of AATD, since the vast majority of such individuals carry the PI*ZZ or PI*SZ genotype, coding for approximately 15% and 25%, respectively, of normal AAT plasma levels. The establishment of international registries, including large series of AATD individuals,5,6 has allowed not only better definition of the epidemiology of AATD, but also more precise definition of the associated clinical phenotypes.5,7,8
Nevertheless, there are at least 30 AAT alleles other than the PI*Z and the PI*S alleles which are associated with significantly reduced or absent plasma AAT levels.9 Given the extreme rarity of such variants, often described in the literature as single case reports, little is known about their epidemiology and even less is known about the associated clinical phenotypes.
The Italian Registry for Severe AATD was established in 1996 as a result of a nationwide screening programme sponsored by the two major Italian scientific respiratory societies.10 Although Italy is considered a country with a medium-low prevalence of AATD (mean PI*Z gene frequency: 0.0013),11 the programme succeeded in identifying a relatively large cohort of AATD individuals. During the development of the screening program, we noticed that, in addition to the groups of AATD individuals carrying the PI*ZZ and PI*SZ genotypes, there was an unexpectedly large group of subjects carrying at least one rare AATD allele. We therefore decided to study this group of subjects, focusing particularly on characterising their clinical phenotypes.
METHODS
Screening programme
The targeted screening programme, based on dried blood spots, has already been described in detail.10 Briefly, paper filters and questionnaires were distributed to respiratory physicians throughout the country. Recommendations for AATD screening were the presence of the following: early-onset COPD, familial clustering of COPD, reduced levels of α1-globulins on electrophoresis, serum levels of AAT <80 mg/dl (nephelometry) or <150 mg/dl (immunodiffusion), or a family history of AATD. Paper filters containing the blood spots were shipped to the Central Phenotyping Laboratory in Rome and submitted to isoelectric focusing. In the case of an abnormal isoelectric focusing pattern, the referring physician was asked to ship a serum sample and a frozen whole blood sample. If the abnormal isoelectric focusing pattern was confirmed, the sample was then investigated at a molecular level. The subject’s demographic and clinical data were retrieved from questionnaires filled in by the referring physicians and shipped together with the specimens. It was recommended that the diagnosis of COPD follow international guidelines.12 All subjects gave their consent to undergo the genetic investigation, which was approved by the ethical committees of the institutions involved.
Key points
-
Most subjects affected by α1-antitrypsin deficiency (AATD) carry the PI*ZZ or PI*SZ genotype. Nevertheless, there are at least 30 AAT alleles other than PI*Z or PI*S associated with reduced or absent plasma AAT levels. Little is known about their epidemiology or associated clinical phenotypes.
-
Over 98 months, 2922 subjects enrolled by the Italian Registry for AATD, which conducts a screening programme based on dried blood spots, were screened. A total of 155 subjects with severe AATD were identified (132 index cases), together with 152 individuals with intermediate AATD (84 cases).
-
Among subjects with severe AATD, we recorded an 11% prevalence of deficient genotypes other than the common PI*ZZ or PI*SZ (15 out of 132 deficient index subjects identified). Among subjects with intermediate AATD, we recorded a 15% prevalence of deficient genotypes other than the common PI*MZ (13 out of the 84 index cases).
-
As the cohort of subjects carrying rare AATD variants was relatively large, we were able investigate their characteristics in terms of associated clinical phenotypes, pulmonary lung function, smoking habit, and geographic distribution. We found that these rare variants had a special position within the spectrum of genotype-phenotype correlations in α1-antitrypsin deficiency.
-
The prevalence of rare α1-antitrypsin variants found in Italy is the highest so far recorded worldwide and, interestingly, occurs in a country with a medium-low prevalence of the common PI*ZZ genotype.
Detection of AATD variants
Genomic DNA was extracted from whole blood cells using the standard technique. The S and Z variants were genotyped by a commercially available amplification-reverse hybridisation test kit (Symbiosis, Cocconato, Italy)13 and by PCR-RFLP using TaqI as the restriction enzyme.14 The genomic DNA was sequenced after PCR amplification of all coding exons (II–V).15 All sequencing products were obtained using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Arrington, UK) and were analysed in an automatic ABI 377 DNA sequencer.
Statistical analysis
Separate files were created for the different genotypes. Subjects homozygous or compound heterozygous for deficiency variants were assigned to the severe AATD group, whereas individuals heterozygous for a deficiency allele and the normal M allele were assigned to the intermediate AATD group. Shapiro-Wilk’s test was used to test the normal distribution for quantitative variables and data values are presented as mean (SD) values. Comparisons between means were performed with analysis of variance, using the Scheffé test for post-hoc comparison. Associations between genotypes and disease status were compared with Fisher’s exact test. To detect associations between FEV1/FVC (forced expiratory volume in 1 s/forced vital capacity), smoke, and genotype, univariate analyses were carried out using logistic regression, followed by multivariate analyses; results are reported as unadjusted and adjusted odds ratios (OR) with 95% confidence intervals (95% CI). A p value <0.05 was considered to indicate statistical significance; all tests were two-sided. Analyses were performed with STATA, release 7.0 (Stata, College Station, TX, USA) and with STATISTICA for Windows (StatSoft, Bedford, UK, 2002).
RESULTS
During the 98 months from February 1996 to April 2004, 2922 subjects were screened, thanks to more than 250 physicians throughout the country who shipped at least one paper filter. A total of 155 individuals (5.3%) with severe AATD were detected (table 1). Of these, 132 were index cases (that is, the probands, constituting 4.5% of the subjects overall), while the other 23 were non-index cases, that is, subjects identified during family screening. Individuals homozygous for the Z allele (PI*ZZ) formed the large majority (114, 74% of the severe AATD; index cases: 96; 73%), followed by a group of 25 individuals (16%) with the compound heterozygous PI*SZ genotype (index cases: 21; 16%). The third group with severe AATD included 16 individuals (10%) carrying at least one rare, non-Z and non-S deficient allele (index cases: 15; 11%). In addition, there was a cohort of 131 subjects with intermediate AATD PI*MZ, accounting for 4.5% of all those screened (index cases: 84; 2.9%) and 21 subjects with intermediate deficiency, resulting from the combination of a rare allele with the normal M variant (0.7% of all those screened; index cases: 13; 0.4%). The rare allele frequency detected was 0.077, taking into account subjects with both severe and intermediate AATD.
Individuals with severe and intermediate AATD identified during the Italian screening programme
The genotyping details of the 37 individuals carrying at least one rare AATD variant are given in table 2. In this series we found 16 individuals carrying the rare variant in a homozygous fashion or in compound heterozygosity with the common AATD variant Z (hereinafter referred to as the rare/deficient (R/D) subset). Twenty one individuals carried the rare variant in heterozygosity with a normal M variant (hereinafter referred to as the M/rare (M/R) subset). The majority of the subjects carried the Mmalton and Mprocida variants. Four subjects carried the Plowell variant, two subjects carried the I variant, and one subject each the Mvarallo, Mheerlen, Q0procida, Q0cairo, and Q0clayton variants.
Details of the genotype in the individuals with rare AAT variants
Demographic details are given in table 3. From now on, we will present data from index cases only. Comparisons were performed among individuals homozygous or compound heterozygous for deficiency alleles (that is, R/D, PI*ZZ, and PI*SZ) and between individuals heterozygous for a deficiency allele and a normal allele (M/R and PI*MZ). There were no differences as far as mean age at diagnosis was concerned. Male subjects were more frequent in the R/D group than in the PI*SZ group. Former smokers were significantly more frequent among R/D individuals than among both PI*ZZ and PI*SZ subjects (p = 0.021 and p = 0.013, respectively). Whole cigarette smoke exposure (current+former) was detected in 100% of R/D individuals v 67% of PI*ZZ subjects and 52% of PI*SZ subjects (p = 0.008 and p = 0.0017, respectively), as well as in 85% of M/R subjects v 52% of PI*MZ subjects (p = 0.03). As far as the plasma AAT levels were concerned, the R/D subset had a lower mean (SD) level of 29 (18) mg/dl, similar to that of the PI*ZZ group (28 (11) mg/dl), but significantly lower than that of the PI*SZ group (62 (16) mg/dl; p<0.0001). The M/R subset had a mean (SD) AAT level of 61 (20) mg/dl, which is significantly lower than that of the related PI*MZ group (93 (23) mg/dl; p<0.0001).
Characteristics of the 132 index subjects with severe AATD (PI*ZZ, PI*SZ, and PI*RD) and the 84 index subjects with intermediate AATD (PI*MZ and PI*MR)
Tables 4 and 5 present the patients’ phenotypes, that is, the associated clinical manifestations and lung function data, retrieved from questionnaires filled in by the referring physicians.
Characteristics of the clinical phenotype
Mean (SD) lung function of individuals with severe and intermediate AATD
Among subjects with severe AATD, COPD (alone or in combination with chronic liver disease) was diagnosed more frequently in the R/D group (100%), than in the two other related groups, PI*ZZ (79%), and PI*SZ (38%, p = 0.004). In seven cases out of 96 PI*ZZ (7%) and in two out of 15 R/D (13%), COPD and liver disease were diagnosed simultaneously. Prevalence of chronic liver disease did not differ among subjects carrying severe AATD; it was absent in M/R subjects, but it was detected in 11% of PI*MZ individuals. A number of other associated conditions were reported: with the exception of one PI*MZ subject diagnosed with Wegener’s granulomatosis, which has been repeatedly associated with AATD,16 the other conditions are most likely to be chance associations. The percentage of healthy subjects increased significantly among the two groups with intermediate AATD (42% in the PI*MZ group, and 46% in the M/R group).
With reference to the functional phenotype (table 5), the mean basal FEV1 value (% predicted) was significantly lower in the R/D group as a whole than in the related PI*SZ group (p = 0.00095), but not significantly lower than in the PI*ZZ group. The mean FEV1/FVC among the five genotype groups was consistently different (table 5). However, when individuals with normal lung function (healthy, liver disease without COPD, other conditions) were disaggregated from the whole group, the FEV1 and FEV1/FVC in the remaining patients with COPD no longer differed among the R/D, PI*ZZ, and PI*SZ subgroups. The difference between PI*MZ and M/R subjects for FEV1 was significant, but it may have been influenced by the low number of M/R subjects with COPD.
The effect of smoking habit and genotype on FEV1/FVC is summarised in table 6. Logistic regression and multivariate analysis showed that smoke and PI*ZZ and PI*RD genotypes act as independent risk factors for an FEV1/FVC<0.70.
Odds ratio and 95% confidence interval (95% CI) for FEV1/FVC<0.70 according to smoking habit and AATD genotype
DISCUSSION
Our finding of a 11% prevalence of subjects with severe AATD carrying genotypes other than PI*ZZ and PI*SZ is, to our knowledge, the highest reported so far. The NHLBI Registry for severe AATD with 1021 subjects includes 1.7% with genotypes other than PI*ZZ or PI*SZ.17 The Alpha One Foundation Research Network Registry also includes subjects with intermediate AATD: individuals with rare AATD variants accounted for 5.7% of the total.18 However, in our series including subjects with both severe and intermediate AATD increased the prevalence to 13%. Thus, our large series of rare AATD variants, detected within the relatively small sized Italian registry, prompted us to investigate the characteristics of these subjects more closely.
Molecular characterisation showed that the majority of subjects with rare AATD variants, including both index and non-index cases, carried at least one Mmalton allele (16/37 individuals, 21/74 alleles). This mutation is raised on the M2 base allele and consists of the deletion of an entire TTC codon in exon II, and subsequent deletion of the Phe51 or Phe52 residue of the mature protein.19,20 Ten of the 44 subjects carried at least one Mprocida allele, based on M1(Val213), a T→C point mutation at codon 41 exon II, leading to a proline for leucine substitution.21 The I allele, found in two individuals, and the Plowell allele (also referred to as QO*Cardiff), found in four individuals, are both raised on the M1(Val213) base allele.20 The I allele is characterised by a C→T point mutation at codon 39 exon II, leading to a cysteine for arginine substitution, whereas the Plowell allele is characterised by an A→T transversion at codon 256 exon III, leading to a valine for asparagine substitution. The Mheerlen variant, found in one subject, is raised on the M1(Ala213) base allele and it is characterised by a C→T point mutation at codon 369 exon V, leading to a leucine for proline substitution.22 The Q0procida allele (also referred to as Nullprocida or Nullisola di procida)23 is a null variant characterised by a 17 kb deletion encompassing exons II–V. The Q0clayton, a null variant raised on the M1(Val213) base allele, more recently identified,24 is characterised by a C insertion in exon V, causing a 3′ frameshift mutation, in turn resulting in a stop codon at residue 376. Finally, two AATD variants were recently found in our series of subjects: the Mvarallo allele was first described in a family from a Northern Italian village. This allele is characterised by a 30 bp deletion accompanied by a 22 bp fragment insertion at the 41–51 codon region in exon II,25 whereas the Q0cairo is a null variant raised on the M1(Val213) base allele, characterised by a A→T transversion at codon 259, exon II, resulting in a stop codon instead of a Lys codon (GenBank accession number AY 256958).
In addition to revealing the high prevalence of rare AAT variants in Italy, which will be discussed later, our relatively large series prompted us to examine the phenotypic profile of carriers of such variants. Taken as a whole and looking at index cases only, there was a higher proportion of men (12/15) and a higher proportion of current or former smokers (100%) among carriers of rare variants than in the other groups with severe AATD (PI*ZZ and PI*SZ) (table 3). Among subjects with intermediate AATD, M/R subjects smoked more than the related PI*MZ subjects (table 3). The higher proportion of former smokers among R/D individuals is intriguing: perhaps they stopped smoking because of the lung disease.
Analysis of COPD prevalence and respiratory function data among the groups revealed interesting findings. We found an increasing prevalence of COPD according to the hierarchy PI*MZ (38%) and PI*SZ (38%)<M/R(46%)<PI*ZZ (79%)<R/D (100%) (table 4). Accordingly, the magnitude of FEV1/FVC ratio impairment followed the same hierarchy: PI*MZ (0.74)<PI*SZ (0.71)<M/R (0.67)<PI*ZZ (0.57)<R/D (0.39) (table 5).
Thus, within the spectrum of prevalence of lung disease associated with AATD, carrying a rare AATD variant(s) places R/D individuals at the highest risk, in the same position as PI*ZZ individuals, whereas M/R individuals are in an intermediate position, with PI*SZ and PI*MZ individuals at the end with lower risk.8,26 Since it is widely accepted that phenotypes result from the interaction between genetic determinants (in this case, the plasma levels of AAT) and environmental determinants (in this case, cigarette smoking), then the phenotypic ranking seems to result from the interaction between the AAT plasma levels reported in our series, with the R/D and PI*ZZ groups at the lowest end (29 and 28 mg/dl, respectively), followed by PI*SZ (62 mg/dl) and M/R (61 mg/dl), and with PI*MZ individuals at the highest end (93 mg/dl), and the smoking prevalence, ranking R/D (100%)>M/R (85%)>PI*ZZ (67%)>PI*SZ (52%) and PI*MZ (52%). These findings further support the concept that the genetic risk factor for COPD is significantly related to the AAT level,1 and that cigarette smoking may influence the risk rate27,28 (table 6).
Chronic liver disease, which is the second most common feature associated with AATD, was detected in 13% of R/D subjects, 13% of PI*ZZ subjects, 19% of PI*SZ subjects, and 11% of PI*MZ subjects (table 4). Diagnosis of chronic liver disease was based on abnormalities in liver function tests; 15% of subjects identified as affected by chronic liver disease had a definitive biopsy-proved diagnosis of liver cirrhosis. The Mmalton variant, the most frequently detected rare AATD variant, is known to be significantly associated, like the common Z deficiency variant, with hepatic disease, due to intracellular accumulation of the AAT protein.1,29 In our series, isolated liver disease was detected in one non-index subject (Z/Plowell) and in association with COPD in two index subjects (Mprocida/Mprocida and Z/Mmalton). Interaction between individual and environmental factors is also likely to influence the development of liver disease in AATD: for example, most of the PI*MZ subjects with liver disease were referred because reduced plasma AAT levels were detected in the presence of alcoholic and/or hepatitis virus infection.4 However, data on liver disease in our series should be taken with caution, for a number of reasons. First of all, it is known that the prevalence of chronic liver disease in AATD increases with age.30 Thus, the prevalence of liver disease in this study might be affected by the relatively young age of the subjects. Secondly, chronic liver disease may be asymptomatic for years and, therefore, the condition might be underestimated at the time of patient assessment by the physicians participating in the screening programme (who are mostly pulmonologists).
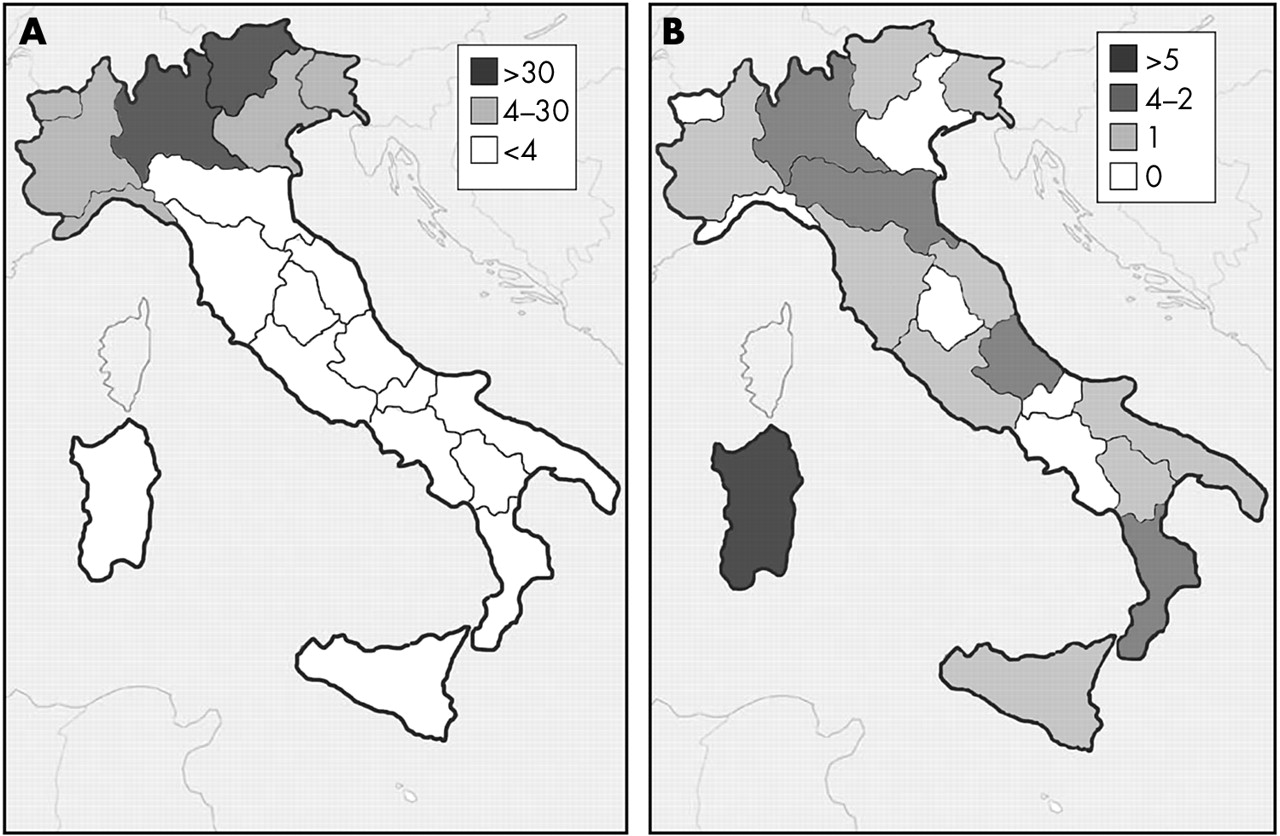
A final point should be made on the geographic distribution of the AATD variants. It is well known that the prevalence of the PI*Z gene in Europe is higher in north-western countries, with a decreasing gradient towards the south-east.3,31 This gradient was respected in Italy, where most PI*ZZ individuals in our series were found in northern areas (fig 1). Interestingly, however, the rare AATD variants displayed a different geographic distribution, peaking in regions, such as Sardinia, with lower PI*ZZ prevalence. Since the nomenclature of many rare AATD variants reflects their probable southern Italian origin (Mprocida, Mpalermo, Q0isola di procida, Q0trastevere), this raises the intriguing question of whether rare AATD variants are more prevalent in areas with a lower PI*Z gene frequency.

{kind=link}
Geographic distribution of index subjects carrying the PI*ZZ genotype (A) and subjects carrying the rare AATD variants (B) enrolled in the Italian Registry for Severe AATD, depicting the range for absolute number of subjects identified for each Italian region. PI*ZZ subjects peak in regions of Northern Italy (Lombardy and Trentino), whereas the rare AATD variants peak in one region of Central Italy (Sardinia).
In conclusion, from the limited data obtained from the Italian Registry for Severe AATD, it seems that individuals carrying rare AATD variants are characterised by a rather peculiar phenotypic profile, placing them in a precise position within the spectrum of genotype-phenotype correlations in AATD. These data, as well as the geographic distribution of rare variants, need verification using larger, international registries.
Acknowledgments
The authors are deeply indebted to all physicians who are participating in the Italian AATD screening program, to members of the Gruppo IDA, to Dr Daniela Medicina, and to Mrs Nuccia Gatta and the Associazione α1-AT. We gratefully acknowledge manuscript editing by Dr Rachel Stenner. ML is a member of the Council of the Alpha One International Registry (A.I.R.).
REFERENCES
Footnotes
-
This study was supported by IRCCS Policlinico San Matteo Ricerca Corrente grants, MIUR Progetti di Interesse Nazionale 2002, the Alpha-1 Foundation, Fondazione Cariplo, Bayer EU, and Altana.
-
Competing interests: none declared