



Clinical characteristics of immunoglobulin G4-positive interstitial pneumonia
- Masamichi Komatsu1,
- Hiroshi Yamamoto1⇑,
- Shoko Matsui2,
- Yasuhiro Terasaki3,
- Akira Hebisawa4,
- Tae Iwasawa5,
- Takeshi Johkoh6,
- Tomohisa Baba7,
- Atsushi Miyamoto8,
- Tomohiro Handa9,
- Keisuke Tomii10,
- Yuko Waseda11,
- Masashi Bando12,
- Haruyuki Ishii13,
- Yasunari Miyazaki14,
- Akihiko Yoshizawa15,
- Tamiko Takemura16,
- Yoshinori Kawabata17 and
- Takashi Ogura7 the Tokyo Diffuse Lung Disease Study Group
- 1First Dept of Internal Medicine, Shinshu University School of Medicine, Matsumoto, Japan
- 2Health Administration Center, University of Toyama, Toyama, Japan
- 3Dept of Analytic Human Pathology, Nippon Medical School, Tokyo, Japan
- 4Division of Clinical Pathology, Asahi Central Hospital, Asahi, Japan
- 5Dept of Radiology, Kanagawa Cardiovascular and Respiratory Center, Yokohama, Japan
- 6Dept of Radiology, Kansai Rosai Hospital, Amagasaki, Japan
- 7Division of Respiratory Medicine, Kanagawa Cardiovascular and Respiratory Center, Yokohama, Japan
- 8Dept of Respiratory Medicine, Respiratory Center, Toranomon Hospital, Tokyo, Japan
- 9Dept of Advanced Medicine for Respiratory Failure, Graduate School of Medicine, Kyoto University, Kyoto, Japan
- 10Dept of Respiratory Medicine, Kobe City Medical Center General Hospital, Kobe, Japan
- 11Third Dept of Internal Medicine, Faculty of Medical Sciences, University of Fukui, Fukui, Japan
- 12Division of Pulmonary Medicine, Dept of Medicine, Jichi Medical University, Tochigi, Japan
- 13Dept of Respiratory Medicine, Kyorin University School of Medicine, Tokyo, Japan
- 14Dept of Respiratory Medicine, Tokyo Medical and Dental University, Tokyo, Japan
- 15Dept of Diagnostic Pathology, Kyoto University Hospital, Kyoto, Japan
- 16Dept of Pathology, Kanagawa Cardiovascular and Respiratory Center, Yokohama, Japan
- 17Division of Diagnostic Pathology, Saitama Cardiovascular and Respiratory Center, Kumagaya, Japan
- Hiroshi Yamamoto (yama5252{at}shinshu-u.ac.jp)
Abstract
This study suggests that IgG4+ IP with abundant IgG4+ cells and elevated serum IgG4 levels could be treated differently from IgG4-related respiratory disease due to potential differences in disease behaviour and response to corticosteroid therapy https://bit.ly/3dUo2cu
To the Editor:
Immunoglobulin G4-related disease is a systemic disease characterised by tumefactive lesions with abundant IgG4-positive plasma cells and elevated serum IgG4 concentrations [1–4]. We previously assessed IgG4-related respiratory disease (IgG4-RRD) with extrathoracic manifestations [5]. IgG4-RRD develops through the lymphatic routes in the lungs and responds well to corticosteroid therapy with a benign prognosis [5–7]. However, whether interstitial pneumonia (IP) with IgG4-positive plasma cell infiltration in the lungs without the extrathoracic lesions of IgG4-related disease could be considered a type of IgG4-RRD is controversial.
Here, we aimed to elucidate the clinico–radio–pathological characteristics of IP with abundant IgG4-positive cells in the lungs and elevated serum IgG4 levels without extrathoracic lesions. We defined this IP as “IgG4-positive IP” in this study.
We recruited patients suspected of having IgG4-positive IP nationwide from March 2019 to May 2019. This study was conducted by the Tokyo Diffuse Lung Disease Study Group. All procedures involving human participants were approved by the Ethics Committee of the Shinshu University School of Medicine (approval number 4465) and the relevant participating institutions. This study was performed in accordance with the Declaration of Helsinki and its subsequent amendments. Additionally, the review board waived the need for patient approval or informed consent because the study involved a retrospective review of patient records.
We reviewed the cases of 28 patients with suspected IgG4-positive IP from 17 institutions throughout Japan. All participants were diagnosed with IP via chest high-resolution computed tomography (HRCT), elevated serum IgG4 concentrations (≥135 mg·dL−1), and infiltrations of abundant IgG4-positive plasma cells (IgG4-positive/IgG-positive cell ratio >40% and IgG4-positive cells >10 per high-power field from the specimens obtained by surgical lung biopsies). HRCT and histopathology patterns of IP were determined according to the 2018 guidelines regarding interstitial pulmonary fibrosis [8] and the 2013 statement regarding idiopathic interstitial pneumonias (IIPs) [9]. Lung specimens were immunostained for IgG and IgG4 in addition to staining with haematoxylin-eosin and Elastica-Masson or Elastica-van Gieson. Patients’ clinical records and radiological and histopathological data were reviewed, and the final diagnoses were established after multidisciplinary discussion (MDD).
Of the 28 patients, 16 were diagnosed with IgG4-positive IP by MDD. Extrathoracic lesions suggesting IgG4-related disease were not present in these 16 patients during the study period. Of the remaining 12 patients, seven patients were diagnosed with IgG4-RRD with extrathoracic lesions, three were diagnosed with multicentric Castleman disease, one had rheumatoid arthritis, and one had lung cancer.
The median age of the 16 patients with IgG4-positive IP at the time of diagnosis was 66 years (range: 49–74 years), and 12 were male. 13 of the 16 patients had a history of smoking. In addition, 13 of the 16 patients had respiratory symptoms, such as cough or dyspnoea, at diagnosis.
Almost all 16 patients had abnormally high serum concentrations of Krebs von Lungen-6 and surfactant protein-D. Contrarily, the white blood cell count and C-reactive protein concentrations were nearly normal. Bronchoalveolar lavage was performed in eight patients; there were no specific trends in the cell fractions of bronchoalveolar lavage fluid. In pulmonary function tests, four patients showed restrictive changes (percentage of predicted vital capacity <80%). The median percentage of the predicted diffusing capacity of the lungs for carbon monoxide was 57.9% (range: 29.9–81.4%).
The most common characteristic findings observed on chest HRCT imaging were ground-glass opacities (n=16), reticular opacities (n=11), traction bronchiectasis (n=10), thickening of the interlobular septal wall (n=9), and hilar mediastinal lymphadenopathy (n=6). Ground-glass and reticular opacities were predominant in the lower and peripheral lung zones. Representative HRCT images are shown in figure 1a. The HRCT patterns found in the 16 patients were as follows: indeterminate for usual interstitial pneumonia (UIP) (n=8), desquamative interstitial pneumonia (DIP) (n=3), unclassifiable IIPs (n=3; two patients had nonspecific interstitial pneumonia (NSIP) with organising pneumonia and one patient had pleuroparenchymal fibroelastosis (PPFE) with UIP), and NSIP (n=2).

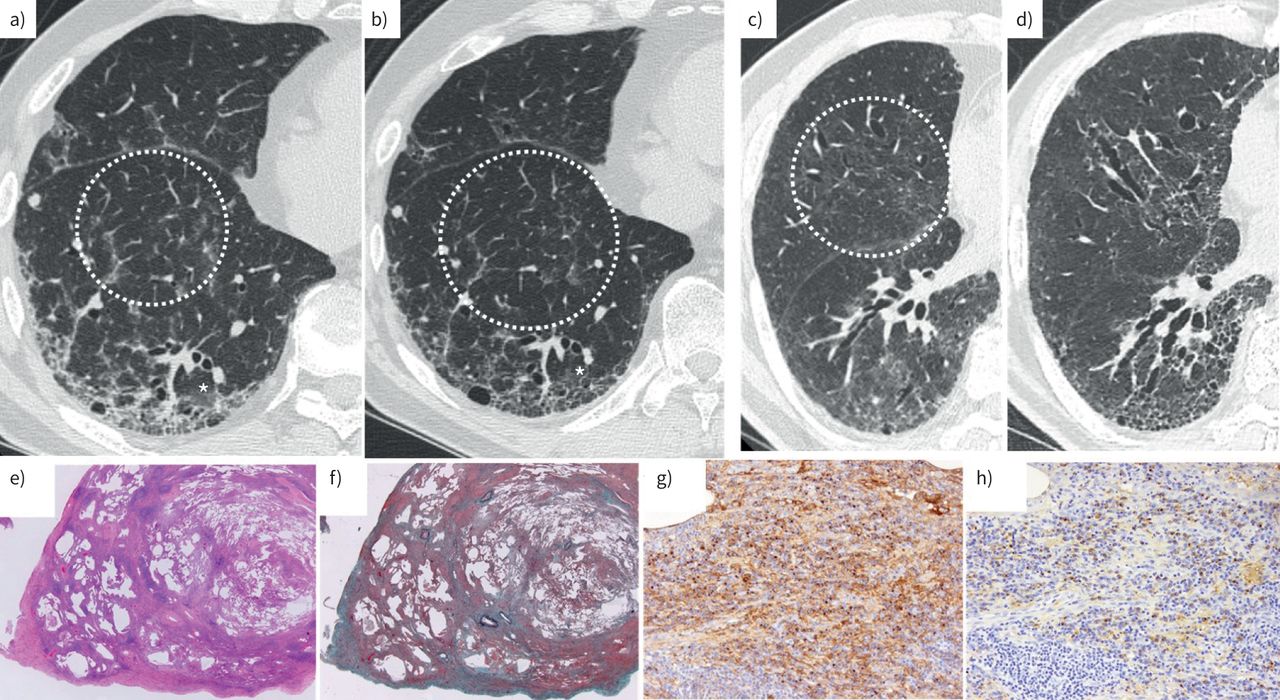{kind=link}
Radiographic and pathological findings of IgG4-positive interstitial pneumonia. a–d) Transverse section of high-resolution computed tomography (HRCT) images of two cases of IgG4-positive interstitial pneumonia. a) Before treatment and b) 2 years after initiation of corticosteroid treatment. The HRCT pattern was indeterminate for usual interstitial pneumonia (UIP). The perilobular ground-glass opacities (circle) improved with corticosteroid treatment; however, reticular opacities remained (asterisk). c) Before treatment and d) 6 years after initiation of corticosteroid treatment. Ground-glass opacities and thickening of the interlobular septal wall (circle) improved with corticosteroid treatment, but traction bronchiectasis and reticular opacities deteriorated. The patient was also treated with an antifibrotic agent, but he eventually died because of an acute exacerbation. e–h) Histopathological features of a representative IgG4-positive interstitial pneumonia case. e, f) Architectural distortion with marked lymphoplasmacytic cell infiltration is seen at a low-power magnification (e) haematoxylin and eosin staining; f) Elastica-Masson staining; magnification 1.25×). The case was of interstitial pneumonia with an unclassifiable pattern because of the combined presentation of usual interstitial pneumonia and nonspecific interstitial pneumonia pattern. g, h) Images showing an increase in the IgG-positive and IgG4-positive cells in the fibrous parenchyma (g) IgG; h) IgG4 immunohistochemistry, respectively; magnification 20×).
Marked numbers of lymphoplasmacytic cells in the fibrous parenchyma were observed in the histopathology of 14 patients. However, none of these patients had either obliterative vasculitis or storiform fibrosis, which are characteristic findings of IgG4-related disease [1–3]. The representative pathological findings are shown in figure 1e–h. IgG4-positive plasma cells were conspicuously present in the fibrous parenchyma. The histopathology patterns of the 16 patients were as follows: unclassifiable IIPs (n=9; four patients had coexisting UIP and NSIP), NSIP (n=3), UIP (n=1), probable UIP (n=1), DIP (n=1), and PPFE (n=1).
The median follow-up period was 59 months (range: 13–126 months). All but one of the 16 patients were treated with corticosteroid monotherapy as initial therapy. With treatment, ground-glass opacities on HRCT improved in all 15 patients (figure 1b). By contrast, the reticular opacities deteriorated after initiation of treatment in six of the 15 patients. The typical HRCT findings before and after treatment are shown in figure 1c–d. The patients who did not receive corticosteroid treatment had a stable clinical course during the follow-up period.
Immunosuppressant and antifibrotic agents were administered as additional therapies to two patients owing to disease progression. Acute exacerbation occurred in two patients. Three patients died during the follow-up period: two patients due to chronic respiratory failure and one patient due to an acute exacerbation.
In this study, we examined the clinico–radio–pathological characteristics of IgG4-positive IP in 16 cases. On HRCT, ground-glass opacities were predominant in peripheral and peribronchovascular areas, and traction bronchiectasis and reticular opacities were observed. Lymphoplasmacytic inflammation into peribronchiolar regions and interlobular septa was seen in many patients. In addition, various types of fibrosis, such as UIP, NSIP, and DIP, were present. We found a correlation between the distribution of ground-glass opacities in HRCT and the lymphoplasmacytic inflammation in the lung tissue. Additionally, a correlation existed between the patterns of traction bronchiectasis and the reticular opacities at HRCT and histopathological fibrosis.
15 of the 16 patients received corticosteroid therapy. Although the ground-glass opacities improved on HRCT for all patients after treatment, reticular opacities, which indicate fibrosis, deteriorated in six patients despite treatment. Furthermore, despite treatment, three patients died during the follow-up period due to disease progression.
Notably, the clinical courses in the study participants differed significantly from that of the previously reported conventional IgG4-RRD [5]. Corticosteroid therapy is fully effective in patients with IgG4-RRD [5–7]. We speculate that the improvement in ground-glass opacities on HRCT reflected the elimination of IgG4-positive lymphoplasmacytic cell infiltration from the perilymphatic stromal area in the surgical lung biopsy specimens. Residual fibrosis may have been the essence of IP, leading to progressive fibrosis, even though the ground-glass opacities improved with corticosteroid treatment.
Some limitations of this study were its retrospective nature and the small sample size, although we did recruit patients with IgG4-positive IP throughout Japan. Further prospective research is necessary to confirm the disease behaviour of IgG4-positive IP.
In summary, we revealed the clinical characteristics of IgG4-positive IP. We believe that IgG4-positive IP needs to be treated as a separate entity from conventional IgG4-RRD because of the relative differences in disease behaviour and responses to corticosteroid therapy.
Acknowledgements
The authors thank Shinichi Sasaki (Juntendo University Urayasu Hospital) and Shinyu Izumi (Central Hospital of the National Center for Global Health and Medicine), who supported the clinical and pathologic review conferences. We also thank Kiminobu Tanizawa (Kyoto University Hospital), Sho Yamada (Kitano Hospital), Machiko Arita, Shinya Yokoe (Kurashiki Central Hospital), Ryosuke Hirabayashi (Kobe City Medical Center General Hospital), Takafumi Yamaya, Masato Asaoka (Kanagawa Cardiovascular and Respiratory Center), Miwa Yamanaka (Shinshu University School of Medicine), Yuhei Ito (Ise Red Cross Hospital), Naoki Hamada (Kyushu University), Toshikazu Takasaki, Hiroyoshi Yamauchi (Jichi Medical University), Nobuhito Arakawa (International University of Health and Welfare Hospital), Hiromi Tomioka (Kobe City Medical Center West Hospital), Kosuke Okuma, Kojiro Honda (Kyorin University School of Medicine), Seiko Takasawa (Tokyo Medical and Dental University), Shun Shinomiya (Saitama Medical University) and Yutaro Nakamura (Hamamatsu University School of Medicine) for case registrations from their respective institutions.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: M. Komatsu, H. Yamamoto, S. Matsui, M. Bando, H. Ishii, Y. Miyazaki and T. Ogura conceptualised the study. M. Komatsu, H. Yamamoto, S. Matsui and T. Ogura designed the study. M. Komatsu, H. Yamamoto, S. Matsui, Y. Terasaki, A. Hebisawa, T. Iwasawa, T. Johkoh, T. Baba, A. Miyamoto, T. Handa, K. Tomii, Y. Waseda, M. Bando, H. Ishii, Y. Miyazaki, A. Yoshizawa, T. Takemura and Y. Kawabata acquired and analysed the data. M. Komatsu performed the statistical analysis and interpreted the data. M. Komatsu, H. Yamamoto, S. Matsui and T. Ogura wrote the manuscript. H. Yamamoto, S. Matsui, Y. Terasaki, A. Hebisawa, T. Iwasawa, T. Johkoh, T. Baba, A. Miyamoto, M. Bando, H. Ishii, Y. Miyazaki and T. Ogura provided administrative support.
Conflict of interest: M. Komatsu has nothing to disclose.
Conflict of interest: H. Yamamoto has nothing to disclose.
Conflict of interest: S. Matsui has nothing to disclose.
Conflict of interest: Y. Terasaki has nothing to disclose.
Conflict of interest: A. Hebisawa has nothing to disclose.
Conflict of interest: T. Iwasawa has nothing to disclose.
Conflict of interest: T. Johkoh has nothing to disclose.
Conflict of interest: T. Baba has nothing to disclose.
Conflict of interest: A. Miyamoto has nothing to disclose.
Conflict of interest: T. Handa has nothing to disclose.
Conflict of interest: K. Tomii has nothing to disclose.
Conflict of interest: Y. Waseda has nothing to disclose.
Conflict of interest: M. Bando has nothing to disclose.
Conflict of interest: H. Ishii has nothing to disclose.
Conflict of interest: Y. Miyazaki has nothing to disclose.
Conflict of interest: A. Yoshizawa has nothing to disclose.
Conflict of interest: T. Takemura has nothing to disclose.
Conflict of interest: Y. Kawabata has nothing to disclose.
Conflict of interest: T. Ogura has nothing to disclose.
Support statement: This study was supported by the Ministry of Health, Labour and Welfare Research Program on Rare and Intractable Diseases (No. JPMH20FC1040), the Japan Society for the Promotion of Science (No. 21K08175), the Tokyo Diffuse Lung Disease Study Group and Shionogi Pharmaceutical, Co., Ltd. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received February 13, 2021.
- Accepted June 23, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org