



Recurrence of primary disease following lung transplantation
- Dorina Rama Esendagli1,7,
- Prince Ntiamoah1,7,
- Elif Kupeli2,
- Abhishek Bhardwaj3,
- Subha Ghosh4,
- Sanjay Mukhopadhyay5 and
- Atul C. Mehta6⇑
- 1Dept of Pulmonary and Critical Care Medicine, Cleveland Clinic, Cleveland, OH, USA
- 2Dept of Pulmonary Diseases, Baskent University, Faculty of Medicine, Ankara, Turkey
- 3Dept of Pulmonary and Critical Care Medicine, Cleveland Clinic, Cleveland, OH, USA
- 4Diagnostic Radiology, Cleveland Clinic, Cleveland, OH, USA
- 5Dept of Pathology, Cleveland Clinic, Cleveland, OH, USA
- 6Dept of Pulmonary and Critical Care Medicine, Cleveland Clinic, Cleveland, OH, USA
- 7Authors contributed equally to this work
- Corresponding author: Atul C. Mehta (mehtaa1{at}ccf.org)
Abstract
Lung transplant has become definitive treatment for patients with several end-stage lung diseases. Since the first attempted lung transplantation in 1963, survival has significantly improved due to advancement in immunosuppression, organ procurement, ex vivo lung perfusion, surgical techniques, prevention of chronic lung allograft dysfunction and bridging to transplant using extracorporeal membrane oxygenation. Despite a steady increase in number of lung transplantations each year, there is still a huge gap between demand and supply of organs available, and work continues to select recipients with potential for best outcomes. According to review of the literature, there are some rare primary diseases that may recur following transplantation. As the number of lung transplants increase, we continue to identify disease processes at highest risk for recurrence, thus shaping our future approaches. While the aim of lung transplantation is improving survival and quality of life, choosing the best recipients is crucial due to a shortage of donated organs. Here we discuss the common disease processes that recur and highlight its impact on overall outcome following lung transplantation.
Abstract
This article reviews the underlying conditions leading to lung transplant with potential for recurrence and the impact of such recurrences on the overall outcome following transplant https://bit.ly/3v3gSvJ
Introduction
Over the past 40 years, lung transplantation (LTx) has become a definitive treatment for patients with a variety of end-stage lung diseases. According to the International Society for Heart and Lung Transplantation (ISHLT), >100 000 LTx have been performed over the years. The 2018 registry report showed the median survival for all adult LTx recipients to be 6.5 years [1]. Lung transplantation should be considered for adults with advanced lung disease who meet the following criteria: 1) high (>50%) risk of death due to lung disease within 2 years if the transplantation is not performed; 2) high (>80%) likelihood of surviving at least 90 days after lung transplantation; and 3) high (>80%) likelihood of 5-year post-transplant survival provided there is adequate graft function [2].
Disease-based indications include idiopathic pulmonary fibrosis (IPF), fibrosing non-specific interstitial pneumonia (NSIP) and other progressive interstitial lung diseases (ILDs) refractory to treatment, ILDs related to collagen vascular diseases (scleroderma, rheumatoid arthritis), COPD, bronchiectasis (cystic fibrosis or non-cystic fibrosis), pulmonary hypertension, α1-antitrypsin deficiency, sarcoidosis, obliterative bronchiolitis, lymphangioleiomyomatosis (LAM), pulmonary Langerhans' cell histiocytosis (PLCH) and retransplantations [3].
Incidentally, a number of conditions can recur following LTx and may involve the transplanted organs. Here, we review conditions leading to transplant with potential for recurrence and the impact of such recurrences on the overall outcome following LTx.
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis is a rare, female-predominant, low-grade neoplastic disorder with a prevalence of two per one million [4]. It is a progressive, cystic lung disease with abnormal proliferation of atypical smooth muscle-like cells and is either sporadic (S-LAM) or related to tuberous sclerosis complex (TSC-LAM) [5]. The disease course of LAM is variable and ranges from mild, stable disease to progressive respiratory failure, with an estimated median survival of over 20 years [6].
Histologically, the LAM cells have both melanoma-related antigens and smooth muscle antigens which are useful for identification [4]. LAM cells possess bi-allelic inactivation of TSC, a tumour suppressor gene that activates the mTOR pathway, which leads to an uncontrolled proliferation and metastasis of LAM cells.
The goal of treatment is mainly the relief of symptoms and management of complications. The MILES study showed that sirolimus treatment could stabilise the function of lung and improve the quality of life [7]. On the other hand, Oprescu et al. [8] in 2013 showed that such therapy does not improve the outcome of disease.
In patients with respiratory failure who have exhausted all medical therapies, LTx may be the only recourse [9]. The first reported lung transplant procedure for LAM was a combined heart–lung transplantation in 1984 [10]. According to 2019 ISHLT data, a total of 582 LTx were performed for LAM between 1995 and 2018 [11, 12].
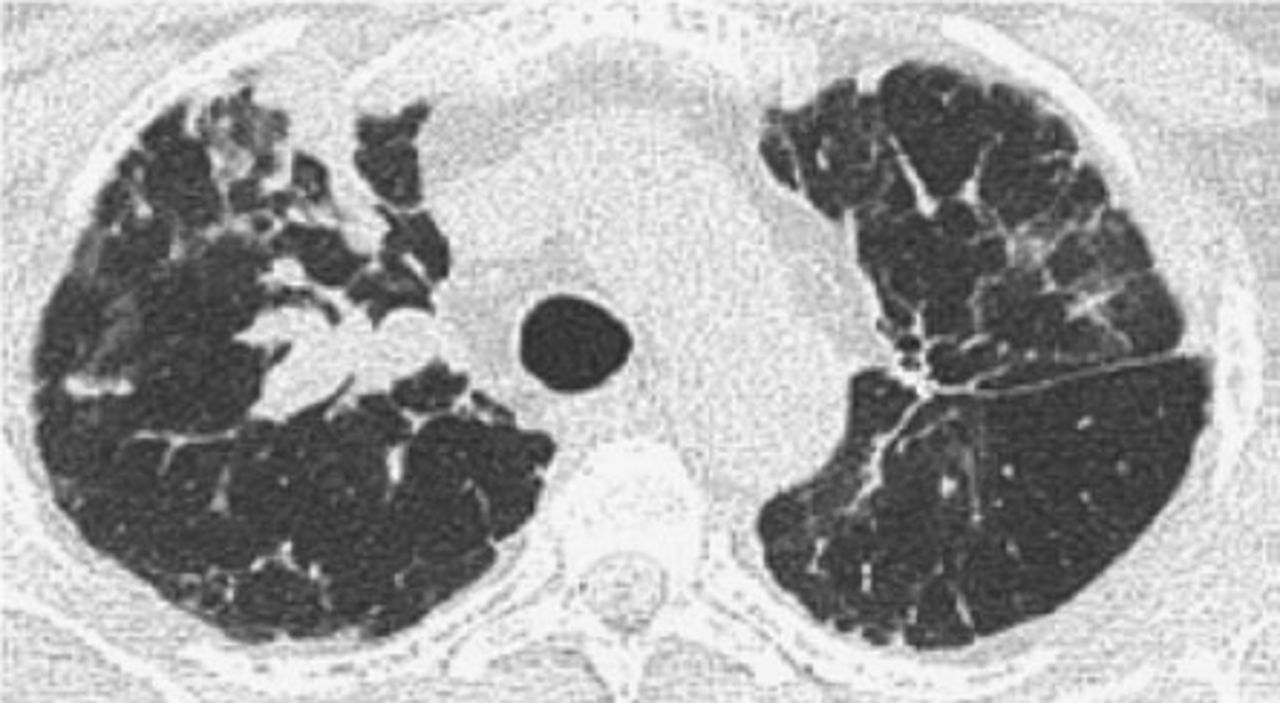
The recurrence of LAM (R-LAM) following LTx is rare and only 23 cases have been reported in the literature. It is evident that LAM could recur as early as within 2 months after LTx (figures 1 and 2). The database from Europe and Japan demonstrated a recurrence rate around 6–7% for LAM after transplantation [13–15]. The recurrence is rare, and the post-transplant survival of these patients when compared to other indications is better and does not compromise long-term survival. The estimated five-year post-transplant survival among LAM patients is between 60% and 70% [13, 14, 16].
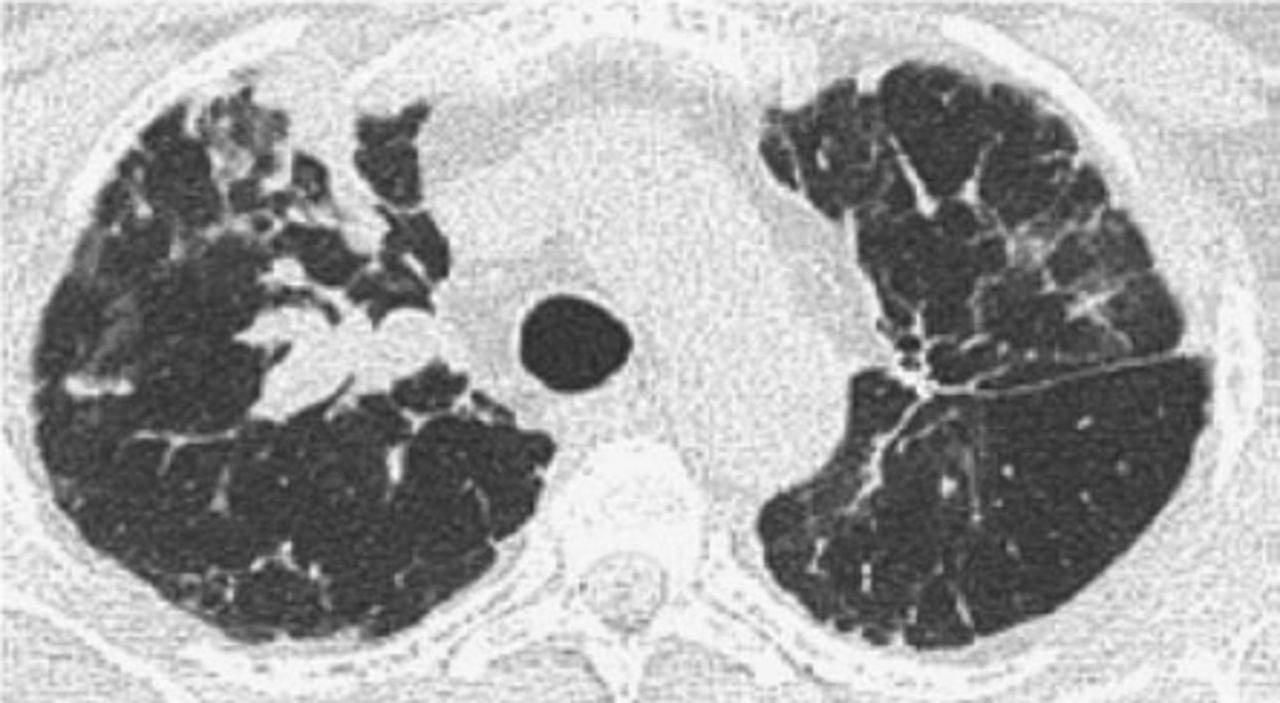
Computed tomography of the chest. Right upper lobe nodules with bilateral interstitial infiltrates and scattered ground–glass opacities proven to be recurrent lymphangioleiomyomatosis following lung transplantation during a surveillance bronchoscopy.
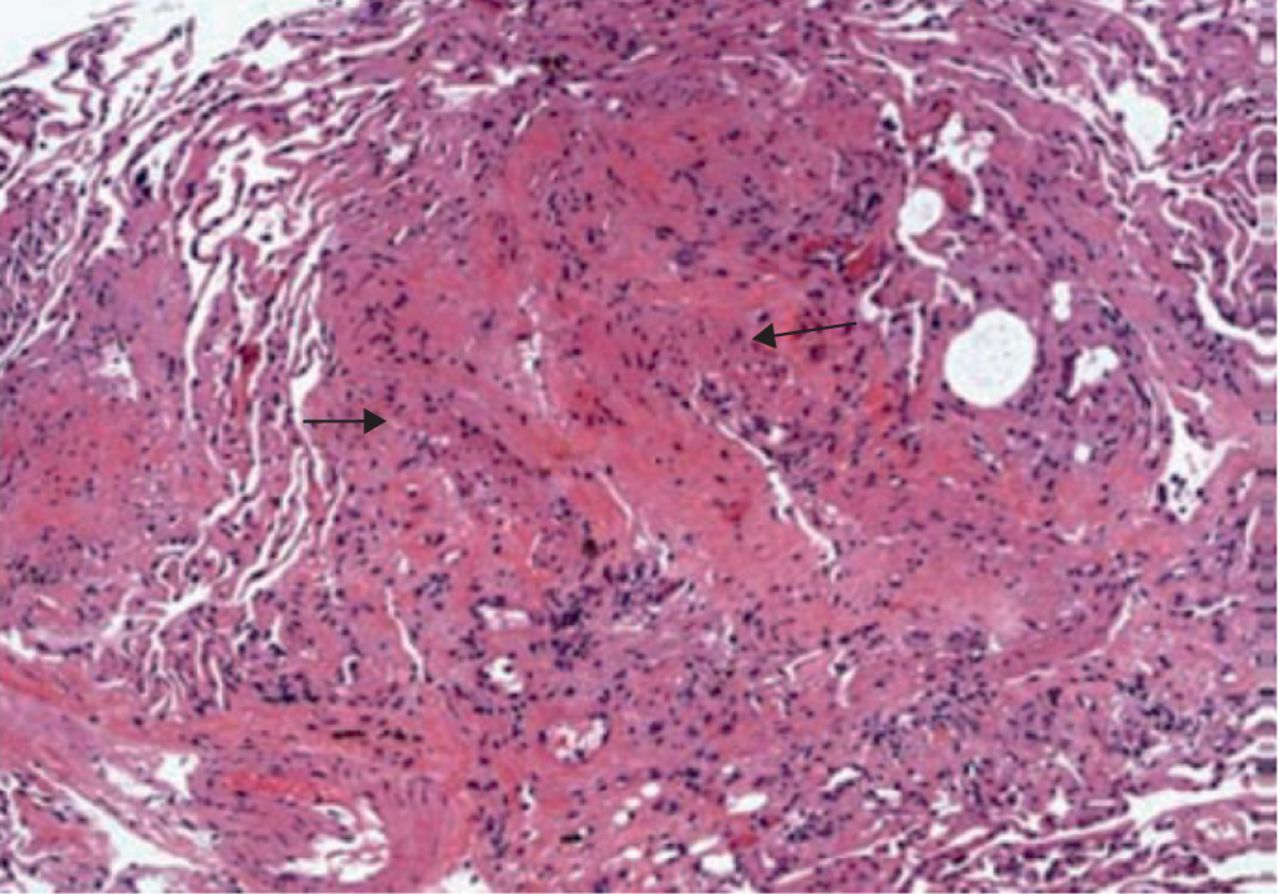
Histopathological examination of the transbronchial biopsy specimen revealing spindle-shaped lymphangioleiomyomatosis cells (arrows) suggestive of recurrence (haematoxylin-eosin, original magnification ×100).
Regarding the possible mechanism of recurrence, genetic analysis by Karbowniczek et al. [5] concluded that LAM cells metastasise to the allograft lung after transplantation, despite their histologically benign features. This is likely facilitated by immunosuppression along with genetic predisposition.
It is not very clear when to start mTOR inhibitors as LAM recurrence is mostly asymptomatic. It is unlikely that a large, randomised trial among these patients is feasible due to its rarity; however, we suggest that in LTx recipients with LAM, sirolimus should be considered as a primary anti-rejection medication either as a mono or as a dual therapy with a calcineurin inhibitor (CNI) [17]. This can be done in the context of CNI-sparing regimens or using a second antiproliferative medication instead of mycophenolate or azathioprine. Whether these would benefit from life-long mTOR-inhibitor therapy remains to be established.
Patients undergoing LTx for LAM have acceptable morbidity and satisfactory survival. The median survival following LTx in LAM is 12 years and better than other lung diseases [18]. It is important to investigate the possibility of disease recurrence post-transplantation in patients who have deteriorating lung function. Intolerance or complications of mTOR inhibitors may limit their use in some patients, who may then require retransplantation. In the literature, only five LAM patients underwent retransplantation [19–21]. Two of them were due to graft failure [20] and bronchiolitis obliterans syndrome [21]. To date, there are no reported cases of LAM patients receiving a transplant for recurrence in the allograft. It is unclear at this stage if mTOR inhibitors or any hormonal therapy can delay or prevent recurrence in the allograft [22–32].
Sarcoidosis
Sarcoidosis is a multisystem disease of unknown aetiology that predominantly affects the lungs and lymph nodes. The histological hallmark of sarcoidosis is the formation of noncaseating epithelioid cell granulomas in affected organs. The T-cell function plays a role in the development of the disease [33, 34]. The prognosis of patients with isolated pulmonary sarcoidosis is generally good. A majority of these patients undergo spontaneous remission within 2 to 5 years. Patients with stage I (according to radiological Scadding stage) disease have over 80% rate of resolution compared with stage II (60%) or stage III (30%) disease. A small number of patients may progress to end-stage lung disease. Mortality ranges from 1 to 6%, with the majority of deaths resulting from respiratory failure [35]. Despite treatment, some individuals develop end-stage lung disease due to parenchymal fibrosis. In stage 3 and 4 patients, disease progression may lead to irreversible destruction and chronic respiratory failure. According to 2019 ISHLT data, a total of 1540 (2.4%) LTx were performed for sarcoidosis between 1995 and 2018 [11]. Indications for LTx in sarcoidosis are functional capacity III and IV, pulmonary hypertension, right atrial pressure of over 15mm Hg and hypoxaemia at rest [12]. Five-year survival following LTx for sarcoidosis is between 47% and 69% [36–38].
Sarcoidosis is the most commonly reported disease to recur post-LTx with an estimated rate of 47% [39]. A recent article reported the recurrence to be 14% thus suggesting that the rate could be influenced by the immunosuppressive regimen which has changed in recent years [38]. Ionescu et al. [40] showed via DNA analysis that recurrence of granulomas in the allografts appears to be of recipient origin. The granulomas appear within the first 6–12 months post-transplant, are usually detected via surveillance biopsies and rarely seem to have a significant impact on allograft function [41, 42]. A case has been reported of sarcoid recurrence after single lung transplantation (SLT), necessitating repeat transplantation, which was followed again by sarcoid recurrence. This report favours bilateral lung transplantation (BLT), which provides a greater functional reserve in case of disease recurrence compared to SLT, and also prevents infections from persistent bronchiectasis [38, 43]. The first case series related to recurrent sarcoidosis was published by Johnson et al. [44] in 1993. The onset of sarcoidosis was seen as early as 2 weeks after the transplantation, mostly within the first 3 months and as late as 2 years [40, 44–46]. The disease tends to be milder in its clinical manifestations than primary disease, possibly due to anti-rejection regimen. Recurrent sarcoidosis may also manifest clinically as either a solitary or numerous miliary nodules [43, 47, 48]. Because the granulomas can be focal and patchy, a negative biopsy does not exclude recurrent sarcoidosis. Normal organs transplanted into recipients with preexisting sarcoidosis are likely to develop sarcoid granulomas, whereas organs from donors with known sarcoidosis transplanted in a non-sarcoid recipient do not appear to develop significant or progressive disease. “Donor-acquired sarcoidosis” is development of sarcoidosis in presumably naïve recipients who have received tissues or organs from donors who were not known or suspected to have active sarcoidosis [40].
The aetiology of sarcoidosis is not known with certainty despite decades-long effort. It is generally thought that it is the result of an exaggerated immune response in a genetically susceptible individual to an undefined antigen, such as certain environmental factors, microbes (e.g. Mycobacterium tuberculosis, Propionibacterium acnes) or partially degraded antigens. Currently, it is believed that both genetic predisposition and environmental factors play essential roles in its pathogenesis [44].
Additionally, in a subclass of patients, an increased usage of the γδ T-cell receptor has been found, which is a similar phenomenon seen in patients with tuberculosis. Mycobacterial DNA was demonstrated in lung cells obtained from patients with sarcoidosis using polymerase chain reaction. The interesting observation of recurring sarcoidosis in patients undergoing LTx may reflect the infectious nature of this disorder, since transmission was observed from donor to recipient as well as from recipient to the donated organ [44, 49, 50].
The decreased occurrence of sarcoidosis relapses from 2013 onwards suggests a role for mTOR inhibitors, which became more widely used to prevent rejection and might inhibit granuloma formation [38, 51, 52]. In addition, there is a possibility that the switch from cyclosporin to tacrolimus in the early 2010s might be implicated in the decreased incidence of relapses, yet the hypothesis remains to be proven [38].
Although recurrence of granulomas in transplanted lungs may occur, this rarely has a significant impact on lung allograft function or recipient survival [41, 53]. The presence of active granulomas on the explanted lung may be a useful predictor of subsequent recurrence. However, recurrence has neither been shown to significantly affect graft function nor worsen the outcomes of these patients. It can easily be treated with systemic corticosteroids. When referring such patients for retransplantation, careful consideration of the benefits and risks of lung transplantation must be made. In a rare and unlikely case of respiratory failure due to recurrent sarcoidosis of the transplanted lung, retransplantation should be assessed on an individual basis and adjustment of the immunosuppressive regimen should be taken into account. Furthermore, extrapulmonary sarcoidosis involvement should be excluded.
Pulmonary Langerhans cell histiocytosis
PLCH is a rare, smoking-related cystic lung disease that can progress to respiratory failure and severe pulmonary hypertension. It is caused by a disorder of myeloid dendritic cells. No occupational or geographic predisposition has been reported, but nearly all affected individuals have a history of current or prior cigarette smoking [54, 55]. PLCH is estimated to account for 3–5% of adult diffuse parenchymal lung diseases. Langerhans cells are normally found in low numbers in the dermis, the reticuloendothelial system, the lung and the pleura. In PLCH, the Langerhans-like cells, which express CD1a, S100 protein, and langerin (CD207), are characteristically found in clusters. Somatic mutations that activate the mitogen-activated protein kinase (MAPK) pathway are present in virtually all cases of Langerhans cell histiocytosis (LCH) and PLCH. In both LCH and PLCH, the most common variants are BRAF V600E and MAPK2K1 genes encoding protein kinases, but numerous others have been described [56]. Smoking promotes accumulation of non-neoplastic CD1a dendritic cells around airways and may also promote maintenance of CD1a cells with oncogenic mutations [56].
Extrapulmonary LCH is noted in <20% of reported cases of PLCH [57]. When present, bone lesions, diabetes insipidus and skin lesions can be seen. PLCH should be suspected in all patients with upper lung zone cystic or nodular radiographic abnormalities, or a history of recurrent pneumothorax, diabetes insipidus or bone pain. A current or past history of smoking or exposure is an important feature. There are no routine laboratory tests that are diagnostic of PLCH [58]. The finding of >5% CD-1a and CD207 positive cells on BAL strongly supports the diagnosis of PLCH [59, 60].
For patients without symptoms or pulmonary impairment, smoking cessation and observation without specific therapy is adequate. The optimal therapy for progressive PLCH has not been determined. Systemic glucocorticoids have a limited role. For patients who are not candidates for or do not respond to glucocorticoids, trial of cladribine or cytarabine is suggested with appropriate monitoring of peripheral blood for cytopenia and prophylaxis against opportunistic infections [61]. Lung transplantation is an option for patients with advanced and progressive PLCH. Precise data regarding prevalence is not available, although a large series of hundreds of patients undergoing surgical lung biopsies for diffuse lung disease reported PLCH in 4–5% of all biopsies [62]. It is an infrequent indication of LTx worldwide, accounting for only 0.4% of adult primary LTx from January 2004 to June 2015 [12]. The recurrence of PLCH after successful lung transplantation is uncommon, with 15 cases in the literature [63–68]. Most of the cases of recurrent disease have been described within 5 to 60 months after transplantation. The first case was published by Gabbay et al. [63], in a 32-year-old nonsmoker male who underwent bilateral LTx and had recurrence of disease after 2 years. Etienne et al. [65] reported two cases of recurrence; both had single LTx. These patients resumed smoking early after transplantation. The recurrence of the disease suggests either that extrapulmonary factors play a role in pathogenesis or the disease may be truly neoplastic. The recent demonstration that Langerhans cells in PLCH may proliferate locally, usually showing an abnormal phenotype, lends some support to the latter theory. Why the disease should recur in exactly the same pulmonary distribution before and after transplantation in some patients is unclear [64].
Dauriat et al. [66] published a multi-centre analysis of 39 patients with PLCH who underwent LTx. Extrapulmonary involvement was present in 31%. The survival rate was 76.9% at 1 year, 63.6% at 2 years, 57.2% at 5 years and 53.7% at 10 years. The recurrence rate was 20% in this patient population with no impact on survival. Three of the patients resumed smoking after LTx. The recurrence rate was significantly higher with extrapulmonary involvement [66]. Furthermore, smoking cessation and corticosteroids are the treatment options for the recurrence. Response to pulse steroid therapy is usually satisfactory, and symptomatic relief can be achieved together with resolution of infiltrates [67].
Unless symptomatic relief is achieved with medical treatment, retransplantation can be considered. Dauriat et al. [66] performed retransplantation on three of 39 patients with PLCH. However, these patients underwent retransplantation for bronchiolitis obliterans syndrome and not for recurrence. In the literature, there are no cases of retransplantation for recurrent PLCH. Nonetheless, the probability of recurrence of the primary disease should be kept in mind, even in patients undergoing retransplantation.
Hard metal exposure
Hard metal lung disease (HMLD) is a rare condition that occurs after chronic occupational exposure to cobalt and tungsten carbide. Giant cell interstitial pneumonia (GIP) is distinct and considered pathognomonic for HMLD, although some cases with no apparent hard metal exposure have been reported. It is different from other occupational lung diseases as it does not depend on the cumulative dosage of the agent [67, 68]. The giant cells seen on the biopsy (figure 3) are referred to as cannibalistic cells that engulf neutrophils and lymphocytes, an uncommon biological process called emperipolesis [69].

{kind=link}
{kind=link}
{kind=link}
a) Note multinucleated giant cell (arrow) within an airspace. The giant cell contains an intact macrophage (arrowhead), a phenomenon known as emperipolesis. The interstitium is thick (asterisk) (haematoxylin-eosin, original magnification ×400). b) The airspaces contain large numbers of multinucleated giant cells (arrows). Note interstitial thickening and a focus of peribronchiolar metaplasia (long arrow). Asterisk: bronchovascular bundle (haematoxylin-eosin, original magnification ×40). c) Several multinucleated giant cells show emperipolesis (white arrows) (haematoxylin-eosin, original magnification ×200). d) Explanted lung showing emperipolesis (arrow). Adjacent interstitium is thickened by fibrosis and chronic inflammation (long arrow) (haematoxylin-eosin, original magnification ×400). e) CD3 shows a few scattered T-lymphocytes (arrowheads) within the interstitium. As expected, multinucleated giant cells are negative for this marker (arrow) (CD3 immunohistochemical stain, original magnification ×200). f) CD68 stain highlights numerous macrophages and giant cells (arrows) (CD68 immunohistochemical stain, original magnification ×100). g) Movat stain highlights collagen deposition within interstitium (Movat pentachrome, original magnification ×200). h) Transplant transbronchial biopsy shows a multinucleated giant cell (arrow) (haematoxylin-eosin, original magnification ×400).
Treatment consists of cessation of exposure, which may facilitate recovery in some patients. However, this is not the option for fibrotic lung disease in which the findings are irreversible. Corticosteroids and other immunosuppressive drugs have been used in some cases, but the efficacy is not yet proven [70, 71]. Although LTx is a choice for end-stage and progressive disease, two cases have been reported with recurrence of the primary disease after transplantation even though the exposure to hard metal was not present [72, 73]. Frost et al. [73] reported a case with single LTx who deteriorated after 2 years. The autopsy showed no evidence of inorganic particles in the allograft but changes typical for GIP were present. Tarabichi et al. [72] reported a case of single LTx for HMLD complicated by recurrent episodes of lung injury and multinucleated cells involving the allograft. There is lack of data concerning the recurrence of HMLD, but according to these case reports an autoimmune mechanism might be responsible for the recurrence as there was no exposure to hard metal in the post-transplant period.
Emphysema due to α1-antitrypsin deficiency
α1-antitrypsin deficiency (AATD) is a genetic disorder with 3.4 million people thought to have this disease worldwide [74]. AAT is secreted mainly by the liver and is key in keeping balance between proteases and antiproteases. It does this by inhibiting pancreatic trypsin, neutrophil elastase, cathepsin G and proteinase-3 [75]. The organs mainly affected by this are the liver and lungs. The condition leads to early onset emphysema with an incidence rate of 1.9% [76, 77]. Smoking is the main trigger related to the development of lung disease especially for the ZZ phenotype.
Management consists of standard treatment for COPD and augmentation therapy with purified pooled human plasma α1-antitrypsin infusion [78]. For patients whose lung function declines despite optimal therapy, LTx can be an option [79].
According to 2019 ISHLT registry data, the total number of LTx for AATD from January 1995 to June 2018 were 2969 (4.7%) of which 2155 were bilateral [11]. The frequency for retransplantation in AATD patients was 11.8% according to Wallinder et al. [19], but the reason for this is not well clarified. There are only two case reports that have shown a recurrence of emphysema after the LTx, and the common reason was the resumption of smoking [80, 81]. Glanville et al. [82] described emphysema on allografts of two AATD patients at post mortem examination, and smoking is the main cause for the recurrence. On the other hand, as this is a genetic disease, even after LTx, the patients may still be at risk for emphysema, but there is not sufficient data for whether augmentation therapy could be of benefit after transplantation. Thus, special attention and rehabilitation for this group is required in order to prevent smoking after transplantation. These observations form the basis for continued replacement therapy post-transplantation.
Pulmonary alveolar proteinosis
Pulmonary alveolar proteinosis (PAP) is a rare disease which involves accumulation of lipoproteinaceous material in the alveolar space because of insufficient clearance of surfactants by the alveolar macrophages [83]. PAP is classified as genetic, secondary or autoimmune, the latter being the most common [83]. Antibodies against granulocyte–macrophage colony-stimulating factor (GM-CSF) is the main reason for the autoimmune type, which further leads to insufficient macrophage clearance of surfactants [84].
The clinical presentation varies from being asymptomatic to dyspnoea, cough, weight loss, chest pain, fatigue, fever and haemoptysis [85, 86]. Periodic acid-Schiff positive eosinophilic granules are present in the foamy macrophages [83].
The main treatment approach is whole lung lavage, which is beneficial in most cases, but a proportion of patients progress to lung fibrosis. Additional therapies like supplemental GM-CSF, rituximab or plasmapheresis have been tried in refractory cases. Lung transplantation is a choice for end-stage disease [87]. There are only three case reports that describe the recurrence of PAP after LTx [88–90].
The first report was by Parker et al. in 1997 [88] that describes a patient with diagnosis of PAP at 27 but had a bilateral LTx at age of 41. She had a relapse of the disease 3 years after lung transplantation. The other two reports include patients with genetic defects who had a recurrence of disease 26 and 16 months after LTx respectively [89, 90]. The underlying genetic defect resulting in persistent pathological macrophages can enable recurrence of the disease by migration of precursor cells from the bone marrow suggesting that LTx should be performed with caution, and bone marrow transplantation might help in this group of patients.
Interestingly PAP can occur in lung allografts of patients with a different primary disease as well. There are case reports of PAP occurrence after LTx performed for IPF, Eisenmerger's syndrome and pulmonary hypertension [91–93]. One case with acute myeloid leukaemia appearing 5 years after the LTx developed PAP after receiving chemotherapy and had a bacterial pneumonia leading to death [94]. These patients were negative for GM-CSF antibodies. Thus secondary PAP due to alveolar injury from either ischaemia, infection or immunosuppression might have played a role in macrophage dysfunction. On the other hand the mechanisms thought to be responsible for PAP development in lung allografts are present in many LTx patients, but only a few of them progress to PAP. Thus further knowledge is needed if there are any other existing additional risk factors.
Interstitial lung diseases
Diffuse parenchymal lung diseases, often collectively called interstitial lung diseases, are a heterogeneous group of disorders classified together due to similar clinical, radiographic, physiological or pathological manifestations. IPF, the most common type of ILD, has the poorest prognosis.
According to the ISHLT registry, ILDs made up almost 21% of the LTx performed from 1995 to 2018 [11]. Even though there is a tendency for BLT, studies have failed to show its superiority over SLT in terms of survival [95, 96]. Another issue is the extrapulmonary involvement of ILDs due to connective tissue disorders (CTD) which may complicate the LTx procedure.
The most common ILD-associated CTDs are scleroderma (61%), rheumatoid arthritis (13%) and polymyositis/dermatomyositis (12%) [96]. Hypersensitivity pneumonitis is another type of ILD caused by inhalation of a specific antigen which can be identified in almost 40% of the cases [97]. The main approach of treatment is to avoid the causative agent, but in some patients the disease progresses to lung fibrosis in spite of this, thus LTx can be a treatment option [98]. Desquamative interstitial pneumonia (DIP) is a rare form of idiopathic interstitial pneumonia in which alveoli are filled with pigmented macrophages [99]. Passive or active smoking, occupational exposure, drug reactions and autoimmune diseases have been reported as causative factors [100]. In recent years the term DIP has been replaced with SRIF (smoking-related interstitial fibrosis). Treatment consists of smoking cessation and systemic corticosteroids, yet it can progress to end-stage disease, requiring LTx [100].
The most common recurrent disease after LTx among ILDs is DIP. Interestingly, King et al. [101] reported recurrence of DIP as early as 1 month after LTx. Verleden et al. [102] and Kotecha et al. [103] reported cases with disease recurrence after 12 and 14 months, respectively. Infections such as Pneumocystis jirovecii pneumonia, cytomegalovirus and Aspergillus pneumonia might have played a role in the recurrence of disease. Recurrence at an early stage resulted in the death of the patient reported by King et al. The two other patients reported by Verleden and Kotecha recovered completely [101–103]. Bhatt et al. reported recurrence of NSIP in a 42 -year-old female patient after BLT [104]. Histology showed accumulation of recipient origin macrophages as early as 2 months post-LTx which progressed to interstitial fibrosis, thus emphasising the importance of host factors for the recurrence of the disorder [104]. Kern et al. [105] reported a single case of hypersensitivity pneumonitis that recurred 3 years after LTx due to ongoing exposure to the causative agent. There is also a case report of recurrence of CTD-related ILD after LTx. It describes a 15-year-old patient diagnosed with polymyositis, unresponsive to immunosuppressive treatment, that underwent BLT after bridging with extracorporeal membrane oxygenation [106]. The patient had recurrence of primary disease with post mortem analysis consistent with pulmonary fibrosis with usual interstitial pneumonia [106]. Most recently, Scallan et al. [107] described a case of recurrence of non-specific interstitial pneumonia of the fibrotic pattern (NSIP-F) in a lung allograft of a patient who underwent BLT for advanced idiopathic fibrotic NSIP (iNSIP-F). It manifested de novo clinical and serological features of antisynthetase syndrome (anti-SS) 30 months post-transplant, which is the first of its kind ever described. A possible explanation for the development of anti-SS in the post-transplant period is the expression of previously cryptic tissue-specific autoantibodies as a result of the single episode of acute rejection 1 month post-transplant despite the apparent lack of connective tissue disease in the donor [107, 108]. Further studies are needed to establish the risk factors for recurrence in ILD patients after LTx.
Idiopathic pulmonary haemosiderosis
Idiopathic pulmonary haemosiderosis (IPH) is a rare condition first described by Virchow in 1864 as “brown lung induration” and is characterised by the clinical triad of haemoptysis, anaemia and pulmonary infiltrates [109]. IPH affects mainly children with 80% of cases in those under 10 years of age [110]. The incidence of IPH is 0.24–1.23 cases per million [111]. The pathogenesis is unknown. Biopsy shows haemosiderin-laden macrophages [112]. Treatment consists of immunosuppressive drugs that include corticosteroids, hydroxychloroquine, azathioprine and cyclophosphamide [112]. LTx can be considered as an option for patients that do not respond to the treatment, develop pulmonary hypertension or have progression to end-stage disease. There are two cases reported regarding the recurrence of IPH after LTx [112, 113]. Owing to rarity of IPH, the outcome of LTx in these patients is yet not very clear and the rate of recurrence needs further investigation. Yet according to the present literature, the outcome of disease even if it recurs after LTx is good and responds well to the immunosuppressive drugs.
Bronchoalveolar carcinoma
Bronchoalveolar carcinoma (BAC) nomenclature has been replaced with adenocarcinoma in situ and minimally invasive adenocarcinoma. Both are indications for referral and listing for LTx if the tumour has a diffuse parenchymal involvement causing respiratory failure or a low quality of life together with unresponsiveness to conventional medical therapies [2]. The recurrence of tumour after resection is especially seen in multifocal disease and the survival is usually not more than 2 years [114]. The poor prognosis and short survival rates have pushed transplantation centres to perform LTx for this group of patients. de Perrot et al. [115]. reported that patients who underwent LTx for diffuse multifocal BAC and survived after the operation (a total of 22 patients) had a recurrence of primary disease as high as 59% in a period of 5 to 49 months (a median of 12 months) Even though the relapse rate was high, the overall disease-free survival for 5 years was around 35% [115]. Another series by Zorn et al. [116] showed a high recurrence rate of the disease (six out of eight patients), but a good survival for the two patients who were disease free and had unrestricted lung function. Another series from Shin et al. [117] reported three out of six patients who underwent LTx for BAC to have recurrence of disease at 10, 39 and 48 months post-transplant. They showed the recurrence to be of recipient origin by analysing the radiological and pathological features of the tumour [117]. Two other studies also showed that the recurrence of disease has similar features to the primary tumour, and the contamination of the allograft with malignant cells from the main airways of the recipient might be the reason. Therefore, better surgical procedures and new techniques are needed to avoid this phenomenon [118, 119]. The high recurrence rate for BAC suggests a revision of the criteria for the LTx referral and listing.
Diffuse panbronchiolitis
Diffuse panbronchiolitis (DPB) was first described in Japan in the 1960s. It is a chronic respiratory disease that affects mainly the bronchioles and can progress to obstructive and suppurative diseases that end up with bronchiectasis [120]. It is characterised by chronic inflammation with mainly lymphocytes, plasma cells, histiocytes, neutrophils and foamy macrophages accumulating around the bronchioles [121, 122]. Macrolides have been shown to be effective in the treatment of DPB, but in patients who do not respond to treatment, or develop pulmonary hypertension and respiratory failure, LTx may be an option [121, 123]. There are two cases of recurrence of the primary disease following LTx for DPB [122–124].
Chronic sinusitis and colonisation with Pseudomonas aeruginosa – commonly seen in DPB patients – might be risk factors for the recurrence of the primary disease; thus, optimal treatment of these two conditions might help for better outcomes.
Pulmonary vascular diseases
Patients with pulmonary vascular diseases that are associated with pulmonary hypertension (PH) without a response to targeted medical therapy are also candidates for LTx [2]. Patients with New York Heart Association (NYHA) Functional Class III or IV despite a trial of at least 3 months of combination therapy including prostanoids, with cardiac index of <2 L·min−1·m−2, mean right atrial pressure of >15 mmHg, 6-min walk test of <350 m, and development of haemoptysis, pericardial effusion and signs of progressive right heart failure are the criteria for listing these patients for LTx [2].
Pulmonary capillary haemangiomatosis, idiopathic pulmonary arterial hypertension (IPAH) and pulmonary veno-occlusive diseases are rare causes of PH that have a poor prognosis and short survival; thus, LTx remains the only definitive treatment choice. According to 2019 ISHLT data, 2.9% of total LTx were performed for IPAH and 1.5% for other forms of PH [11]. There are only four case reports of recurrence following LTx [125–128]. According to these case reports, the recurrence of such diseases is within 1 year and raises the question of whether LTx is an appropriate treatment choice for this group of patients. The gaps in our knowledge regarding the pathogenesis of these diseases and the possible extrapulmonary involvement warrants further study.
Conclusion
According to the analysis of the present literature about the recurrence of primary diseases after LTx, it is obvious that it is a rare entity and rarely associated with worse outcome. There are gaps in our knowledge regarding the pathophysiology, systemic involvement and risk factors for recurrence on an individual basis, and this has potential to influence criteria for lung transplant listings. Risk factors for recurrence should be elucidated and taken into account to further optimise long-term outcome of patients at risk. Furthermore, recurrence of disease should be considered in new-onset allograft dysfunction, which should be excluded before the diagnosis of chronic lung allograft rejection is established. While the aim of LTx is to prolong survival and quality of life for patients with end-stage lung disease, it is crucial to choose the best recipients due to shortage of donated organs.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00038-2022.SUPPLEMENT
Footnotes
Provenance: Submitted article, peer reviewed.
This article has supplementary material available from https://openres.ersjournals.com/
Conflict of interest: None declared.
- Received March 11, 2022.
- Accepted April 12, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org